Hepatocellular carcinoma
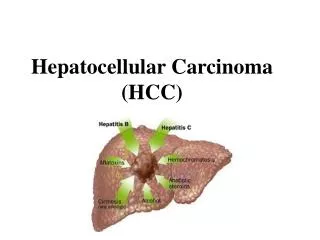
Hepatocellular carcinoma. Hepatocellular carcinoma (HCC) is a primary malignancy of the hepatocyte , derived from well-differentiated hepatocytes , generally leading to death within 6-20 months.
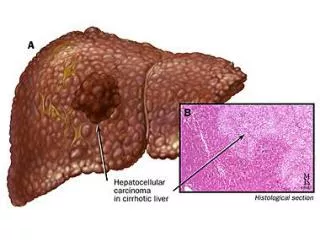

Hepatocellular carcinoma
E N D
Presentation Transcript
Hepatocellular carcinoma • Hepatocellular carcinoma (HCC) is a primary malignancy of the hepatocyte, derived from well-differentiated hepatocytes, generally leading to death within 6-20 months. • Frequently arises in the setting of cirrhosis, appearing 20-30 years following the initial insult to the liver. • 25% of patients have no history or risk factors for the development of cirrhosis.
Epidemiology • HCC is the fifth most common cancer and the third most frequent cause of cancer death worldwide, with an estimated 560,000 new cases per year. • There are strong etiologic associations with chronic hepatitis B virus (HBV), chronic hepatitis C virus (HCV), alcoholic cirrhosis, other causes of chronic liver disease, and dietary aflatoxinexposure. • In patients with underlying cirrhosis, males greatly outnumber females but in non-cirrhotic cases this sex difference is less striking. • In areas of high incidence the peak age is in the third and fourth decades of life but in Europe and North America most cases occur in the fifth and sixth decades.
ETIOLOGY AND RISK FACTORS • Chronic hepatitis B • Chronic hepatitis C • Dietary aflatoxin exposure • Cirrhosis from alcohol and other chronic liver diseases • Familial and genetic influences on risk for hepatocellular carcinoma
Chronic hepatitis B • Cirrhosis develops in 0.1% to 2% of patients with chronic hepatitis B each year, depending on the duration of HBV replication, the severity of disease, and the presence of coexisting infections or alcohol use. • Certain HBV strains predominate in HCCs, suggesting that particular genotypes are more effective at initiating or promoting hepatocarcinogenesis.
Chronic hepatitis C • Genotype 1b is associated with more severe liver disease • Patients with active hepatitis, those with co-infectionswith HBV or human immunodeficiency virus (HIV) and those with other causes of chronic liver disease such as alcoholic liver disease are more likely to develop liver-related complications. • Patients with combined hepatitis B surface antigen positivity and chronic HCV infection are at particular risk for HCC. • After surgical resection of HCC, the presence of active hepatitis and hepatitis C viremia are risk factors for tumor recurrence.
Dietary aflatoxin exposure • Aflatoxin (derived from aspergillusflavustoxin) is a mycotoxin produced by two fungal species, AspergillusflavusandAspergillusparasiticus. • There are at least 13 different types of aflatoxin, with aflatoxinB1 considered the most toxic. • Aflatoxins are metabolized in the liver by the cytochrome P450 and glutathione S-transferase enzyme systems. • AflatoxinB1 is a procarcinogen that is converted in the liver to the mutagenic metabolite aflatoxin B1-8,9-exo-epoxide by hepatic microsomalcytochrome P450.
Cirrhosis from alcohol and other chronic liver diseases • The HCC risk is higher for individuals with chronic hepatitis and ongoing hepatocellular injury. • Individuals who stop drinking significantly reduce their risk for HCC. • Patients with nonalcoholic steatohepatitis and cirrhosis who achieve reduced hepatic steatosis and those with hereditary hemachromatosis and cirrhosis who are iron depleted by phlebotomy have a reduced risk for HCC. • Certain causes of cirrhosis, such as autoimmune hepatitis, carry a relatively low risk for HCC.
Familial and genetic influences on risk for hepatocellular carcinoma • The biologic basis for this phenomenon is unknown. • Epidemiologic studies have suggested either the presence of a recessive allele that contributes to risk for HCC or a familial predisposition to a prolonged HBV replication phase. • Genetic polymorphisms of the carcinogen-metabolizing enzymes cytochrome P450 (CYP), glutathione S-transferase (GST) M1 and N-acetyltransferase(NAT2), as well as p53 polymorphisms, may contribute to familial risk for HCC.
The molecular mechanisms of liver carcinogenesis (1) the use of chemical tumor initiators and promoters in animal models (2) studies of growth factors and their signaling pathways (3) transgenic mouse models overexpressing cytokines, growth factors or oncogenes (4) studies of immune-mediated mechanisms of hepatocellularinjury (5) analysis of the molecular genetic changes that occur in HCCs, including studies of chromosomal allelic imbalance, comparative genomic hybridization, restriction landmark genomic scanning and gene expression analysis using cDNAmicroarrays (6) studies of the molecular consequences of HBV integration and the interaction of the protein products of HBV and HCV with host cell processes
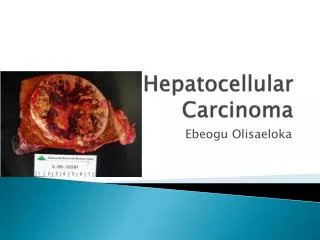
Macroscopic • The photo above shows a view of a longitudinal slice taken through the full length of the liver. The closer view, shows tumor at the top, cirrhotic liver at the bottom and a fibrous reaction in between. • Hepatocellular carcinomas can have a variety of gross patterns, including multinodular/multifocal, such as this one.
Histology • Histology is quite variable, ranging from well-differentiated tumors to anaplastic tumors. • The fibrolamellar subtype is associated with a better prognosis, possibly because it is not associated with cirrhosis and is more likely to be resectable. • The presence of intracellular bile or staining for AFP may be helpful in distinguishing hepatocellular carcinoma from other hepatic malignancies (eg, cholangiocarcinoma). • Immunohistochemistry using the marker Hep-Par 1 may aid in the diagnosis. • Aberrations of chromosome 1 and 8 are common features of hepatocellular carcinoma that can be detected by fluorescent in situ hybridization (FISH) technique. The role of FISH in the diagnosis of hepatocellular carcinoma is still under investigation.
Symptoms • HCC initially may escape clinical recognition because it occurs in patients with underlying cirrhosis and the symptoms and signs may suggest progression of the underlying disease. • decompensation of cirrhosis • tumoral symptoms • cholestasis • fever of unclear etiology • paraneoplastic phenomena • metastatic presentation
Decompensation of cirrhosis • ascites • refractory varicealbleeding the diffuse infiltrating cancers further worsen hepatic function, resulting in: • hyperbilirubinemia • coagulopathy • encephalopathy
Hepatic carcinoma, primary. Dilated collateral superficial abdominal veins in a 67-year-old man with cirrhosis, hepatocellular carcinoma (HCC), and portal vein occlusion.
Tumoral symptoms - less common than the presentation of hepatic decompensation • anorexia • weight loss • general malaise • painful hepatomegaly - the pain is usually acute, limited in time (less than 24 hours) and assumed to be biliary colic
Cholestasis • tumor-associated extrinsic compression of the large bile ducts • tumor invasion of the large bile ducts • miliarymetastasis throughout the liver • hemobiliaand gastrointestinal bleeding • disproportionately high serum transaminases in addition to hyperbilirubinemia and jaundice
Fever • unusual presentation of HCC • the febrile episodes may be discrete episodes and associated with leukocytosis • night sweats are common in advanced HCC and portend a poor prognosis
Paraneoplastic phenomena • are rare • hypercalcemia • erythrocytosis • hypoglycemia • thrombophlebitismigrans • arterial hypertension • the syndrome of watery diarrhea, hypokalemiaand achlorhydria
Metastatic presentation • one metastatic complication of HCC is dyspneadue to tumor pulmonary emboli • another is headache associated with pituitary metastases • Common sites for HCC metastases include the diaphragm with associated pleural effusions, bone metastases with pain and regional lymph node metastases, which may be associated with abdominal discomfort. • Abdominal pain in patients with HCC is a poor prognostic symptom and usually indicates advanced disease.
Physical findings • Jaundice • Ascites • Hepatomegaly • Alcoholic stigmata (Dupuytren contracture, spider angiomata) • Asterixis • Pedal edema • Periumbilical collateral veins • Enlarged hemorrhoidal veins
Differential diagnosis • Cholangiocarcinoma • Cirrhosis • Hepatocellular adenoma • Dysplastic nodules in cirrhosis • Fibrous nodular hyperplasia • Metastatic disease • Primary hepatic lymphoma • Hepatoblastoma • Hemangioma
Haematological and biochemical indices • Apart from alpha-fetoprotein, are non-specific and reflect the space-occupying lesion as well as the underlying cirrhosis present in about 80% of cases • Alpha-fetoprotein is a glycoprotein synthesized by the fetal liver and its plasma concentrations reach their maximum at the end of the first trimester (3 to 4 mg/ml) and then decline. • Raised levels are found in about 80% of patients with hepatocellular carcinoma • Concentrations above 500 ng/ml in a patient with liver disease are highly suggestive • Other tumour markers for hepatocellular carcinoma have been described in the serum, including an abnormal vitamin B 12 binding protein which is usually present with the fibrolamellar histological variant.
Results consistent with cirrhosis: • total bilirubin • aspartateaminotransferase (AST) • alkaline phosphatase • albumin • prothrombin time
Real-time ultrasound • This is a sensitive and specific test and picks up hepatocellular carcinoma in 85 to 90% of cases. • False-negative results usually occur in patients with tumours of less than 2 cm in diameter. • Is frequently used to screen high-risk populations and should be the first test if HCC is suspected.
Abdominal computed tomographic (CT) scanning • This technique is probably no more accurate in detecting hepatocellular carcinoma than ultrasound and should be reserved for cases in which doubt persists. • Sensitivity can be increased by contrast enhancement. • Dynamic spiral contrast-enhanced CT scanning is even more sensitive.
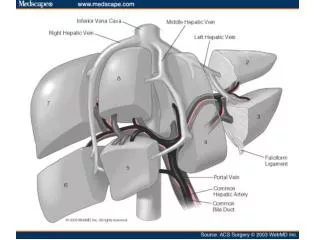
Hepatic arteriography • As the major vascular supply to a hepatocellularcarcinoma is usually arterial, diagnostic changes are seen in a high proportion of cases. • Information gained on the anatomical distribution of the tumour and the vascular anatomy is essential if surgical resection or transplantation is being contemplated and consideration can also be given at the time of arteriography to intra-arterial chemotherapy and hepatic artery embolization. 64-year-old woman with hepatocellular carcinoma. Celiac arteriogram shows standard hepatic artery anatomy.
Magnetic resonance imaging (MRI) • This technique, particularly with the addition of a contrast agent, is proving useful in identifying and characterizing focal liver masses. • Lesions that do not contain reticuloendothelial cells or hepatocytes (haemangiomas and metastases) do not have their signal intensities altered.
Liver biopsy • For definitive diagnosis, liver biopsy is essential, although this is not always possible because of the prolongation of the prothrombin time. • The diagnosis can be considered highly likely without liver biopsy proof if the alpha-fetoprotein level is greater that 500 ng/ml and the hepatic arteriogram shows a tumourcirculation. • Biopsy may be conveniently done at the time of laparoscopy or ultrasonography and suspicious areas can be sampled under direct vision. • Because of the risk of tumourspread, biopsy is often avoided if curative resection or transplantation is planned.
Laparoscopy • To permit liver biopsy under direct vision. • This approach has the additional advantage of sometimes identifying patients who have a localized resectabletumor suitable for partial hepatectomy.
Staging of hepatocellular carcinoma for selection of therapy • all patients should undergo a chest radiograph to exclude pulmonary metastases and at least two imaging studies to stage intrahepatic disease and exclude vascular invasion • ultrasound • contrast CT • contrast MRI • color-flow Doppler and ultrasound-guided FNA (portal vein thrombosis)
Other exams • Chest radiography may demonstrate pulmonary metastases • Bone scanning and head CT scanning are of low yield in the absence of specific symptoms. • PET scan has been evaluated in the experimental setting, but, to date, its role is uncertain. Routine use of PET scan for diagnosis or staging of hepatocellular carcinoma is not recommended.
Hepatic carcinoma, primary. Unusual location of a bone metastasis from hepatocellular carcinoma (HCC).
TNM • T1 - Solitary tumor without vascular invasion • T2 - Solitary tumor with vascular invasion or multiple tumors none more than 5 cm • T3 - Multiple tumors more than 5 cm or tumor involving a major branch of the portal or hepatic vein(s) • T4 - Tumor(s) with direct invasion of adjacent organs other than the gallbladder or with perforation of visceral peritoneum • N0 - Indicates no nodal involvement • N1 - Indicates regional nodal involvement • M0 - Indicates no distant metastasis • M1 - Indicates metastasis presence beyond the liver
Stage grouping • Stage I = T1 + N0 + M0 • Stage II = T2 + N0 + M0 • Stage IIIA = T3 + N0 + M0 • Stage IIIB = T4 + N0 + M0 • Stage IIIC = TX + N1 + M0 • Stage IVB = TX + NX + M1
CLIP scoring system: Score of 0-2 is assigned for each of the 4 features listed below; cumulative score ranging from 0-6 is the CLIP score. • Child-Pugh stage • Stage A = 0 • Stage B = 1 • Stage C = 2 • Tumor morphology • Uninodular and extension less than 50% = 0 • Multinodular and extension less than 50% = 1 • Massive and extension greater than 50% = 2 • Alpha-fetoprotein • Less than 400 = 0 • Greater than 400 = 1 • Portal vein thrombosis • Absent = 0 • Present = 1 - Estimated survival based on CLIP score: Patients with a total CLIP score of 0 have an estimated survival of 31 months; those with score of 1, about 27 months; score of 2, 13 months; score of 3, 8 months; and scores 4-6, approximately 2 months.
Complications • Symptoms of hepatic failure may signify tumor recurrence and/or progression.
Screening • Patients with an increased risk of developing hepatocellular carcinoma, such as those with cirrhosis or chronic HBV or HCV infection, should be considered for regular screening by alpha-fetoprotein assay and abdominal ultrasonography.
Prognosis • This is a highly malignant tumourand the mean survival in most series is around 4 to 6 months. • Patients with cirrhosis have a poorer prognosis than those without. • Encapsulated tumours and the fibrolamellar histological variant, as well as small tumours picked-up at screening, have a better prognosis.
Essentials • Jaundice is the clinical sign of hyperbilirubinaemia, and hence usually indicates disease of the liver or biliary tree. • The pigment in the tissues in best seen as yellowing of the sclera; eventually the skin and soft palate become tinted, but not saliva or sputum. • The urine usually becomes dark. • Rarely, carotenaemia, from eating excessive carrots or vitamin A, can mimic jaundice, but its colour is more prominent in the palms than the sclera.
Definition • Jaundice (icterus) is the yellow discoloration of skin, sclera and mucous membranes caused by the excessive accumulation of bilirubin pigments. • It is usually apparent when bilirubin levels exceed 3 mg/dL or 50 μmol/L.