Download
1 / 30
300 likes | 466 Views
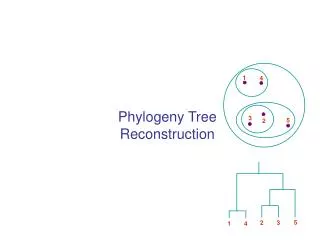
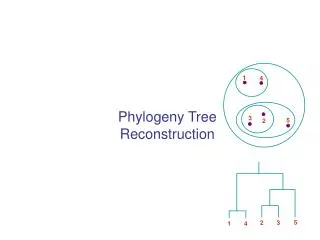
1. 4. 3. 5. 2. 5. 2. 3. 1. 4. Phylogeny Tree Reconstruction. Inferring Phylogenies. Trees can be inferred by several criteria: Morphology of the organisms Sequence comparison Example: Orc: A C A G T G A C G CCCC AAA C G T Elf: A C A G T G A C G C T A C AAA C G T

E N D
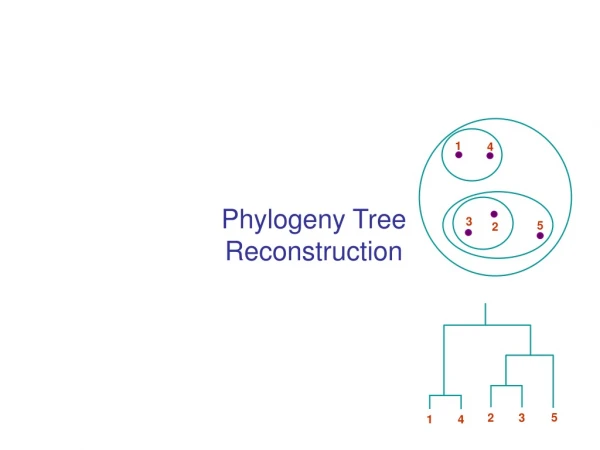
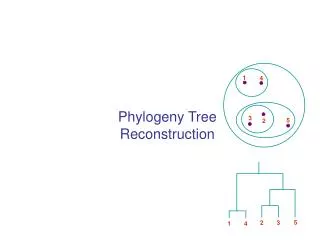
1 4 3 5 2 5 2 3 1 4 Phylogeny Tree Reconstruction
Inferring Phylogenies Trees can be inferred by several criteria: • Morphology of the organisms • Sequence comparison Example: Orc: ACAGTGACGCCCCAAACGT Elf: ACAGTGACGCTACAAACGT Dwarf: CCTGTGACGTAACAAACGA Hobbit: CCTGTGACGTAGCAAACGA Human: CCTGTGACGTAGCAAACGA
Modeling Evolution During infinitesimal time t, there is not enough time for two substitutions to happen on the same nucleotide So we can estimate P(x | y, t), for x, y {A, C, G, T} Then let P(A|A, t) …… P(A|T, t) S(t) = … … … … P(T|A, t) …… P(T|T, t) x x t y
Modeling Evolution A C Reasonable assumption: multiplicative (implying a stationary Markov process) S(t+t’) = S(t)S(t’) That is, P(x | y, t+t’) = z P(x | z, t) P(z | y, t’) Jukes-Cantor: constant rate of evolution 1 - 3 For short time , S() = I+R = 1 - 3 1 - 3 1 - 3 T G
Modeling Evolution Jukes-Cantor: For longer times, r(t) s(t) s(t) s(t) S(t) = s(t) r(t) s(t) s(t) s(t) s(t) r(t) s(t) s(t) s(t) s(t) r(t) Where we can derive: r(t) = ¼ (1 + 3 e-4t) s(t) = ¼ (1 – e-4t) S(t+) = S(t)S() = S(t)(I + R) Therefore, (S(t+) – S(t))/ = S(t) R At the limit of 0, S’(t) = S(t) R Equivalently, r’ = -3r + 3s s’ = -s + r Those diff. equations lead to: r(t) = ¼ (1 + 3 e-4t) s(t) = ¼ (1 – e-4t)
Modeling Evolution Kimura: Transitions: A/G, C/T Transversions: A/T, A/C, G/T, C/G Transitions (rate ) are much more likely than transversions (rate ) r(t)s(t)u(t)s(t) S(t) = s(t)r(t)s(t)u(t) u(t)s(t)r(t)s(t) s(t)u(t)s(t)r(t) Where s(t) = ¼ (1 – e-4t) u(t) = ¼ (1 + e-4t – e-2(+)t) r(t) = 1 – 2s(t) – u(t)
Parsimony • One of the most popular methods Idea: Find the tree that explains the observed sequences with a minimal number of substitutions Two computational subproblems: • Find the parsimony cost of a given tree (easy) • Search through all tree topologies (hard)
Parsimony Scoring Given a tree, and an alignment column u Label internal nodes to minimize the number of required substitutions Initialization: Set cost C = 0; k = 2N – 1 Iteration: If k is a leaf, set Rk = { xk[u] } If k is not a leaf, Let i, j be the daughter nodes; Set Rk = Ri Rj if intersection is nonempty Set Rk = Ri Rj, and C += 1, if intersection is empty Termination: Minimal cost of tree for column u, = C
Example {A, B} C+=1 {A} {A, B} C+=1 B A B A {B} {B} {A} {A}
Example {B} {A,B} {A} {B} {A} {A,B} {A} A A A A B B A B {A} {A} {A} {A} {B} {B} {A} {B}
Number of labeled unrooted tree topologies • How many possibilities are there for leaf 4? 2 1 4 4 4 3
Number of labeled unrooted tree topologies • How many possibilities are there for leaf 4? For the 4th leaf, there are 3 possibilities 2 1 4 3
Number of labeled unrooted tree topologies • How many possibilities are there for leaf 5? For the 5th leaf, there are 5 possibilities 2 1 4 5 3
Number of labeled unrooted tree topologies • How many possibilities are there for leaf 6? For the 6th leaf, there are 7 possibilities 2 1 4 5 3
Number of labeled unrooted tree topologies • How many possibilities are there for leaf n? For the nth leaf, there are 2n – 5 possibilities 2 1 4 5 3
Number of labeled unrooted tree topologies • #unrooted trees for n taxa: (2n-5)*(2n-7)*...*3*1 = (2n-5)! / [2n-3*(n-3)!] • #rooted trees for n taxa: (2n-3)*(2n-5)*(2n-7)*...*3 = (2n-3)! / [2n-2*(n-2)!] 2 1 N = 10 #unrooted: 2,027,025 #rooted: 34,459,425 N = 30 #unrooted: 8.7x1036 #rooted: 4.95x1038 4 5 3
Probabilistic Methods A more refined measure of evolution along a tree than parsimony P(x1, x2, xroot | t1, t2) = P(xroot) P(x1 | t1, xroot) P(x2 | t2, xroot) If we use Jukes-Cantor, for example, and x1 = xroot = A, x2 = C, t1 = t2 = 1, = pA¼(1 + 3e-4α) ¼(1 – e-4α) = (¼)3(1 + 3e-4α)(1 – e-4α) xroot t1 t2 x1 x2
Computing the Likelihood of a Tree xk • Define P(Lk | a): probability of subtree rooted at xk, given that xk = a • Then, P(Lk | a) = (bP(Li | b) P(b | a, tki))(cP(Lj | c) P(c | a, tki)) tkj tki xj xi
Felsenstein’s Likelihood Algorithm To calculate P(x1, x2, …, xN | T, t) Initialization: Set k = 2N – 1 Recursion: Compute P(Lk | a) for all a If k is a leaf node: Set P(Lk | a) = 1(a = xk) If k is not a leaf node: 1. Compute P(Li | b), P(Lj | b) for all b, for daughter nodes i, j 2. Set P(Lk | a) = b, cP(b | a, ti)P(Li | b) P(c | a, tj) P(Lj | c) Termination: Likelihood at this column = P(x1, x2, …, xN | T, t) = aP(L2N-1 | a)P(a)
Probabilistic Methods Given M (ungapped) alignment columns of N sequences, • Define likelihood of a tree: L(T, t) = P(Data | T, t) = m=1…M P(x1m, …, xnm, T, t) Maximum Likelihood Reconstruction: • Given data X = (xij), find a topology T and length vector t that maximize likelihood L(T, t)
ML Reconstruction • Task 1 • Edge length estimation • Given a topology, find the edge lengths that maximize the likelihood • Accomplished by iterative methods such as Expectation Maximization (EM) or using Newton’s Raphson optimization • Each iteration requires computations that take on the order of the number of taxa times the number of sequence positions • Guaranteed to find local maxima but, in practice, usually find the global maximum
ML Reconstruction • Task 2 • Find a tree topology that maximizes the likelihood • More challenging • Naïve, exhaustive search of tree space is infeasible • Effectiveness of heuristic paradigms, like simulated annealing, genetic algorithms or other search methods is hampered by re-estimating edge lengths afresh for different trees • Problem is tackled by iterative procedures that greedily construct the desired tree
Structural EM Approach • Based on development of Structural EM method in learning Bayesian networks • Similar to standard EM procedure, with exception that we optimize not only edge lengths but also the topology during each EM iteration • Overview to Algorithm • Choose a tree (T1, t1) using, say, Neighbor-Joining • Improve the tree in successive iterations • In the l’th iteration, we start with the bifurcating tree (Tl, tl) and construct a new bifurcating tree (Tl+1, tl+1). Basically we use (Tl, tl) to define a measure Q(T,t:Tl, tl) of expected log likelihood of trees and then to find a bifurcating tree that maximizes this expected log likelihood
Definition of Terms • Σ – fixed, finite alphabet of characterse.g. Σ = {A,C,G,T} for DNA sequences • T – bifurcating tree, in which each internal node is adjacent to exactly three other nodes • N – number of sequences/nodes in the tree • Xi – random variable representing the node i in the tree T, where i = 1,2,..,N • (i,j) Є T – nodes i and j in T have an edge between them • t – vector comprising of a non-negative duration or edge length ti,j for each edge (i,j) Є T • The pair (T,t) constitutes a phylogenetic tree
Definition of Terms • D – complete data set • pa->b(t) • Probability of a character to tranform from ‘a’ into ‘b’ in duration t • Equal to Σc pa->c(t) pc->b(t’) where c ЄΣ • pa - prior distribution of character ‘a’ • Si(a) • frequency count of occurrences of letters • It is the number of times we see the character ‘a’ at node i in D • Si(a) = Σm 1{Xi[m] = a} • Si,j(a,b) • frequency count of co-occurrences of letters • It is the number of times we see the character ‘a’ at node i and the character ‘b’ at node j in D • Si,j(a,b) = Σm 1{Xi[m] = a, Xj[m] = b}
Structural EM Iterations • Three steps • E-Step: Compute expected frequency counts for all links (i,j) and for all character states a,b ЄΣ • M-Step: Optimize link lengths by computing for each link (i,j), its best length • M-Step II: • Construct a topology T*l+1 that maximizes Wl+1(T), by finding maximizing spanning tree • Construct a bifurcating topologyTl+1 such that L(T*l+1, tl+1) = L(Tl+1,tl+1)
E-Step • For each iteration calculate expected frequency count for all links (i,j) using the formula,E[Si,j(a,b) |D, Tl, tl] = Σm P(Xi[m] = a, Xj[m] = b | x[1…N][m], Tl,tl) • Here x[1…N][m]= the set of characters observed at all nodes at position ‘m’ in the data • (Tl,tl) is the current topology • Computation of these expected counts takes O(M.N2|Σ|3) and storing them requires O(N2|Σ|2 ) space • We can improve the speed by O(|Σ|) by calculating approximate counts instead S*i,j(a,b) =Σm P(Xi[m] = a| x[1..N][m], Tl,tl) P(Xj[m] = b | x[1..N][m], Tl,tl)
M-Steps • M-Step I • After each E-Step, we calculate each link (i,j)’s best length using, tl+1i,j= argmaxt Llocal(E[Si,j(a, b) | D, Tl, tl], t), where Llocal(E[Si,j(a, b) | D, Tl, tl], t) = Σa,b E[Si,j(a,b)] [log pa->b(t) – log pb] • M-Step II • Construct a topology T*l+1 that maximizes Wl+1(T), by finding maximizing spanning tree • Construct a bifurcating topologyTl+1 such that L(T*l+1, tl+1) = L(Tl+1,tl+1)
Empirical Evaluation • The Algorithm has been written in C++ and been implemented in a program called SEMPHY • For protein sequences • Compared results to MOLPHY, which is a leading ML application for phylogeny reconstruction based on protein sequences • Used the basic Structural EM algorithm, using a Neighbor-joining tree as a starting point • Evaluated performance on synthetic data sets, generated by constructing an original phylogeny and then sampling from its distributions • The synthetic data sets were broken into a test set and training set • Ran both programs on the data
Empirical Evaluation • For protein sequences: • Results of basic Structural EM • Quality of both methods deteriorated when number of taxa increases and when the number of training position decreases. In this case, both methods did poorly • SEMPHY outperforms MOLPHY in terms of quality of solutions • MOLPHY’s running time grows much faster than SEMPHY’S running time, which is only quadratic in number of taxa • Both MOLPHY’s and SEMPHY’s running times grows linearly in number of training positions