Download
1 / 43
430 likes | 517 Views
Explore how molecular properties are connected to chemical structure and learn about quantum mechanics in drug discovery. Understand electron distribution and wave function in predicting molecular behaviors.

E N D
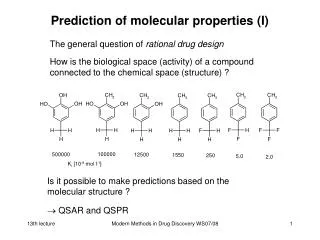
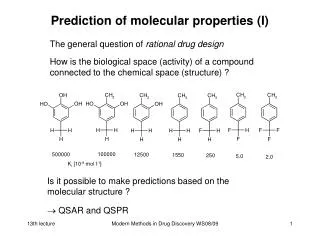
Prediction of molecular properties (I) The general question of rational drug design How is the biological space (activity) of a compound connected to the chemical space (structure) ? Is it possible to make predictions based on the molecular structure ? QSAR and QSPR Modern Methods in Drug Discovery WS08/09
Prediction of molecular properties (II) What are molecular properties? molecular weight MW (from the sum formula C12H11N3O2) melting point boiling point vapour pressure solubility (in water) charge dipole moment polarizability ionization potential electrostatic potential observables Directly computable from the electronic wave function of a molecule Modern Methods in Drug Discovery WS08/09
Prediction of molecular properties (III) All molecular properties that can be measured by physico-chemical methods (so called observables) can also be computed directly by quantum chemical methods. Required: A mathematical description of the electron distribution e.g. by the electronic wave function of the molecule Electron distribution Quantum mechanics (QM) Molecular mechanics (MM)force fields Atomic coordinates Modern Methods in Drug Discovery WS08/09
Quantum mechanics (I) To make the mathematical formalism practically useable, a number of approximations are necessary. One of the most fundamental consists in separating the movement of the atomic cores from that of the electrons, the so called Born-Oppenheimer Approximation: Atomic cores are > 1000 times heavier than the electrons und thus notice the electrons only as an averaged field The (electrostatic) interaction between charged particles (electrons, cores) is expressed by Coulomb‘s law Modern Methods in Drug Discovery WS08/09
Quantum mechanics (II) As electrons are particles, their movement can be described by classical mechanics according to Newtons 2nd law: As electrons are also very small particles (quanta), they exhibit properties of particles as well as those of waves: particle wave galvanic diffraction precipitation Modern Methods in Drug Discovery WS08/09
Schrödinger equation (I) Electrons can be described in the form of a wave function by the time-dependent Schröder equation If the Hamilton operator H is time-independent, the time-dependence of the wave function can be separated as a phase factor, which leads to the time-independent Schrödinger equation. Here, only the dependence from the coordinates remains. Modern Methods in Drug Discovery WS08/09
The wave function (I) The wave function is a mathematical expression describing the spacial arrangement of the (fluctuating) electrons. The squared wave function holds the propability P to find the particle (electron) at a given place in space. P is a so-called observable, whereas the wave function itself is no observable, physical quantity. Thus, integration over the complete space must yield 1 (= total propability to find the electron somewhere in space). Modern Methods in Drug Discovery WS08/09
The Hamilton operator The Hamilton operator contains the kinetic (T) and the potential (V) energy operators of all considered particles i in the system with the squared Nabla operator with As a consequence of the Born-Oppenheimer approximation, also the Hamilton operator can be separated into a core and an electronic part. Modern Methods in Drug Discovery WS08/09
The wave function (II) Any mathematical expression of the wave function must fulfill certain criteria to account for the physical nature of the electrons. As a simplification the wave function of all electrons in a molecule is assumed to be the product of one-electron functions which themselves describe a single electron. • These function must obey some rules: • electrons are indistinguisable • they repell each other • the Pauli principle (two electrons with different spin can share a common state (orbital)) Modern Methods in Drug Discovery WS08/09
Schrödinger equation (II) According to the Schrödinger equation there must be several different energetic levels for the electrons within an atom or molecule. These (orbital) energies can be obtained by integration and rearrangement to The resulting energies are, however, dependend on the quality of the applied wave function and thus always higher or, in the best case, equal to the actual energy. In the simplest case we chose 1s orbitals as basis set to describe the wave function Modern Methods in Drug Discovery WS08/09
Molecular Orbital Theory (I) Molecular orbitals can be constructed as a linear combination of atomic orbitals (LCAO approach) or other basis functions. e.g. for H2 Common expression for a MO with the atomic orbitals Modern Methods in Drug Discovery WS08/09
Molecuar Orbital Theory (II) Applying the LCAO approach for the wave function we yield for H2 • • =1 =1 overlap intergral S • Due to the normalization of the wave function regarding the complete space: Modern Methods in Drug Discovery WS08/09
Molecular Orbital Theory (III) Common notation of the Sekular equations using matrices: The solutions of these Sekular equations for E yield the energies of the bonding and anti-bonding MOs The main numerical effort consists in the iterative search for suitable coefficients (cA, cB, ...) that produces reasonable orbital energies variational principle Hartree-Fock equations Self Consistent Field (SCF) method Modern Methods in Drug Discovery WS08/09
Hückel Theory (I) • (1931) Limited to planar, conjugated -systems,-orbitals are neglected. • The original aim was to interpret the non-additive properties of aromatic compounds (e.g. benzene compared to “cyclohexatrien”) regarding their heats of combustion. The -orbitals are obtained as linear combinations of atomic orbitals (LCAO of pz-orbitals). The -electrons move in an electric field produced by the -electrons and the atomic cores. Modern Methods in Drug Discovery WS08/09
Hückel Theory (II) • Example: ethene H2C=CH2 Modern Methods in Drug Discovery WS08/09
Hückel Theory (III) Within the Hückel theory the Fock matrix contains as many columns, respectively rows, as atoms are present in the molecule. All diagonal elements correspond to an atom i and are set to the value . Off-diagonal elements are only non-zero if there is a bond between the atoms i and j. This resonance parameter is set to (<0). Values for can be obtained experimentally from UV/VIS-spectra ( -4.62 eV). Example butadiene: 1 2 3 4 1 2 3 4 Modern Methods in Drug Discovery WS08/09
Hückel Theory (IV) For a cyclic -system as in benzene, the orbital energies and orbital coefficients results to This also yields the Hückel rule:a system of [4n+2] -electrons is aromatic. Modern Methods in Drug Discovery WS08/09
Hückel Theory (V) • Application of the Hückel method to predict and interpret UV/VIS spectra • Different parameters for different atoms (C,N,O) allow the application of the Hückel theory to further compounds • Orbital energies can be determined experimentally by photo electron spectroscopy (PES) and thus also (the respective ionization potential) and Modern Methods in Drug Discovery WS08/09
Hartree-Fock based methods HY = EY Born-Oppenheimer approximation one-determinant approach Hartree-Fock-equations ZDO-approximation valence electrons parameters optimized basis sets all electron RHF semiempirical methods with minimal basis set ab initio methods with limited basis set multi-determinant approaches Valence electrons UHF ECP spin (a,b) space semiempirical C.I. methods CI MCSCF CASSCF Modern Methods in Drug Discovery WS08/09
Semiempirical methods (I) The problem of ab initio calculation is their N4 dependence from the number of two-electron integrals. These arise from the number of basis functions and the interactions between electrons on different atoms. In semiempirical methods the numerical effort is strongly reduced by assumptions and approaches: 1. Only valence electrons are considered, the other electrons and the core charge are described by an effective potential for each atom (frozen core). 2. Only a minimal basis set is used (one s and three p-orbitals per atom), but using precise STOs that are orthogonal to each other. 3. More or less stringent use of the Zero Differential Overlap (ZDO) approach. Modern Methods in Drug Discovery WS08/09
Semiempirical methods (II) Since 1965 a series of semiempirical methods have been presented from which still some are in use today for the simulation of electromagnetic spectra: CNDO/S, INDO/S, ZINDO Following methods have shown to be particularly successful in predicting molecular properties: MNDO (Modified Neglect of Diatomic Overlap) Thiel et al. 1975, AM1 (Austin Model 1) Dewar et al. 1985 und PM3 (Parameterized Method 3) J.P.P. Stewart 1989 This is partly also due to their availability of the wide spreadMOPAC program package and its later commerical sucessors. All three method are based on the same NDDO approach and differ in the parameterization of the respective elements. Modern Methods in Drug Discovery WS08/09
Non commerical programs MOPAC 7.1 (and MOPAC2007) J.J.P. Stewart http://openmopac.net/ GHEMICAL http://www.bioinformatics.org/ghemical/ghemical/index.html Modern Methods in Drug Discovery WS08/09
AM1 (Austin Model 1) Dewar, Stewart et al. J.Am.Chem.Soc.107 (1985) 3902 Advantages compared to MNDO: + better molecular geometries esp. for hypervalent elements (P, S) + H-bonds (but with a tendency towards forking) + activation energies for chemical reactions • Deficiencies of AM1 (and all other methods based on NDDO): • - hypervalent elements in general, because no d-orbitals • compounds with lone electron pairs (esp. anomeric effect) • NO2 containing compounds Modern Methods in Drug Discovery WS08/09
PM3 (Parameterized Method 3) J.J.P. Stewart J.Comput.Chem.10 (1989) 209 Parametrization was performed more rigerously using errror minimization than in previous methods. Advantages compared to AM1: + better molecular geometries for C, H, P and S + NO2 containing compounds better • Disadvantages compared to AM1: • All other nitrogen containing compounds worse • higher atomic charges lead to a more polar character of the molecules • Not all parameterized elements (e.g. Mg and Al) yield reliable results for all substance classes Modern Methods in Drug Discovery WS08/09
Molecular properties from semiempirical QM calculations (I) • In contrast to ab initio calculations the semiempirical methods MNDO, AM1, and PM3 were calibrated to reproduce experimental data: • heats of formation [Bildungswärmen] • molecular geometries (bond lengths, bond angles) • dipole moments • ionization potentials The results of semiempirical methods regarding these properties are therefore often better than that of ab initio calculations at low level (with comparable computational effort) Modern Methods in Drug Discovery WS08/09
Heats of formation Computation of heats of formation at 25° C atomizationenergies Heats of formation of the elements Experimentally known Only the electronic energy has to be computed Modern Methods in Drug Discovery WS08/09
Comparison of the methods Calculated heats of formation at 25° C for different compoundsAverage mean error (in kcal/mol) • Number of compounds method • (C, H, N, O, and) MNDO AM1 PM3 MNDO/d • Al (29) 22.1 10.5 16.4 4.9 • Si (84) 12.0 8.5 6.0 6.3 • P (43) 38.7 14.5 17.1 7.6 • S (99) 48.4 10.3 7.5 5.6 • Cl (85) 39.4 29.1 10.4 3.9 • Br (51) 16.2 15.2 8.1 3.4 • I (42) 25.4 21.7 13.4 4.0 • Zn (18) 21.0 16.9 14.7 4.9 • Hg (37) 13.7 9.0 7.7 2.2 • Mg (48) 9.3 15.4 12.0 9.3 Modern Methods in Drug Discovery WS08/09
New semiempirical methods since 1995 MNDO/d Thiel & Voityuk J.Phys.Chem.100 (1996) 616 Expands the MNDO methods by d-obitals and is “compatible” with the other MNDO parameterized elements PM3(tm), PM5 d-orbitals for transition elements (transition metals) SAM1 Semi ab initio Method 1 Certain integrals are thouroghly computed, therefore also applicable to transition metals (esp. Cu and Fe) AM1* Winget, Horn et al. J.Mol.Model.9 (2003) 408.d-orbitals for elements from the 3rd row on (P,S, Cl) Modern Methods in Drug Discovery WS08/09
Electronic molecular properties (I) Besides the structure of molecules all other electronic properties can be calculated. Many of those result as response of the molecule to an external disturbance:Removal of one electron ionization potential In general a disturbance by an electric field can be expressed in the form of a Taylor expansion. In the case of an external electrical field F the induced dipole moment mind is obtained as: mo permanent dipol moment of the molecule (if present) a polarizability b (first) hyperpolarizability Modern Methods in Drug Discovery WS08/09
Electronic molecular properties (II) Selection of properties that can be computed from the n-th derivative of the energy according to external fields electr. magn. nuc.spin coord. property 0 0 0 0 energy1 0 0 0 electric dipol moment0 1 0 0 magnetic dipol moment0 0 1 0 hyperfine coupling constant (EPR)0 0 0 1 energy gradient (geom.optimization)2 0 0 0 electric polarizability3 0 0 0 (first) hyperpolarizability0 0 0 2 harmonic vibration (IR)1 0 0 1 IR absorption intensities1 1 0 0 circular dichroisms (CD)0 0 2 0 nuclear spin coupling const. (NMR)0 1 1 0 nuclear magnetic shielding (NMR) Modern Methods in Drug Discovery WS08/09
Molecular electrostatic potential (I) Due to the atomic cores Z and the electrons i of a molecule a spacial charge distribution arises. At any point r the arising potential V(r) can be determined to: While the core part contains the charges of the atomic cores only, the wave function has to be used for the electronic part. Remember: In force fields atomic charges (placed on the atoms) are used to reproduce the electric multipoles and the charge distribution. Modern Methods in Drug Discovery WS08/09
Molecular electrostatic potential (II) To determine the MEP at a point r the integration is practically replaced by a summation of sufficientlysmall volume elements. For visualization the MEP is projected e.g. onto the van der Waals surface. Other possibilities are the representation of surfaces with the same potential (isocontour) From: A. Leach, Molecular Modelling,2nd ed. Modern Methods in Drug Discovery WS08/09
Molecular electrostatic potential (III) Knowledge of these surface charges enables computation of atomic charges (e.g. for use in force fields) ESP derived atomic charges These atomic charges must in turn reproduce the electric multipoles (dipole, quadrupole,...).Therefore the fitting procedures work iteratively. literature: Cox & Williams J.Comput.Chem.2 (1981) 304 Bieneman & Wiberg J.Comput.Chem.11 (1990) 361CHELPG approach Singh & Kollman J.Comput.Chem.5 (1984) 129RESP approach atomic charges for the AMBER force field Modern Methods in Drug Discovery WS08/09
Quantum mechanical descriptors (selection) atomic charges (partial atomic charges) No observables ! Mulliken population analysis electrostatic potential (ESP) derived charges dipole moment polarizability HOMO / LUMO energies of the frontier orbitals given in eV WienerJ (Pfad Nummer) covalent hydrogen bond acidity/basicity difference of the HOMO/LUMO energies compared to those of water Lit: M. Karelson et al. Chem.Rev.96 (1996) 1027 Modern Methods in Drug Discovery WS08/09
Molecular properties from semiempirical methods (II) Which method for which purpose ? structural properties only (molecular geometries): PM3 esp. for NO2 compounds, otherwise AM1 electronic properties: MNDO for halogen containing compounds (F, Cl, Br, I) AM1 for hypervalent elements (P,S), H-bonds Do not mix descriptors computed from different semiempirical methods !e.g. PM3 for NO2 containing molecules and AM1 for the remaining compounds in the set. Modern Methods in Drug Discovery WS08/09
Prediction of Molecular Properties, Examples Descriptors from semiempirical methods(ionization potential, dipole moment ...)along commonly used variables in QSAR equations and classification schemes.Often much more qualitative experimental data than quantitative date are available. • in vitro mutagenicity of MX compounds • Blood-brain distribution (logBB) • CNS permeability of substances • QT-interval prolongation (hERG channel blockers) Modern Methods in Drug Discovery WS08/09
Quantum QSAR Generation of molecular properties as descriptors for QSAR-equations from quantum mechanical data. Example: mutagenicity of MX compounds ln(TA100) = -13.57 E(LUMO) –12.98 ; r = 0.82 Lit.: K. Tuppurainen et al. Mutat. Res.247 (1991) 97. Modern Methods in Drug Discovery WS08/09
BBB-model with 12 descriptors Descriptors mainly from QM calculations: electrostatic surface, principal components of the geometry,H-bond properties Lit: M. Hutter J.Comput.-Aided.Mol.Des. 17(2003) 415. Modern Methods in Drug Discovery WS08/09
96% mde34 95% ar5 CNS Permeability CNS– CNS+ 91% vxbal qsum+ 96% 100% 99% hlsurf 82% qsum+ 100% 99% qsum+ 100% qsumo 72% 100% qsum– 88% 100% 100% pcgc 100% qsum– mpolar 79% 100% 100% 89% 77% cooh dipdens 83% 100% qsum– 89% 100% 80% hbdon 100% size & shape 99% qsum+ 100% 86% dipm electrostatic 100% kap3a 89% 100% H-bonds mde13 92% 94% Modern Methods in Drug Discovery WS08/09 Lit.: C.Andres & M.Hutter QSAR Comb.Sci.25 (2006) 305. 96% 100%
90% 89% mghbd Decision tree for QT-prolonging drugs 100% size & shape 88% chbba electrostatic 100% hlsurf 75% MR 89% 73% hy 100% H-bonds 96% MR hacsurf mde23 99% 100% 96% 100% mpolar 86% 71% 100% sgeca 95% logP 100% MR 89% SMARTS 100% qsumn dipdens 87% 100% 92% 100% 82% t1e MR 83% 100% QT+ QT– 88% t2e 100% 99% logP 100% 93% MR Level of accuracy in % 100% Modern Methods in Drug Discovery WS08/09 qsumn 96% 100% 90% 89% mghbd 100% 88% chbba 100% hlsurf 75% MR 89% QT+ QT– 73% hy 100% 96% MR hacsurf mde23 99% 100% 96% 100% mpolar 86% 71% 100% sgeca 95% logP 100% MR 89% SMARTS 100% qsumn dipdens 87% 100% 92% 100% 82% t1e MR 83% 100% 88% t2e 100% 99% logP 100% 93% MR 100% qsumn 96% 100%
Common structural features of QT-prolonging drugs Derived common substructure expressed as SMARTS string Lit.: M.Gepp & M.Hutter Bioorg.Med.Chem.14 (2006) 5325. Modern Methods in Drug Discovery WS08/09
Molecular properties from force fields As as principal consequence force fields show an even more emphasized dependence from the underlying parameterization. Thus only predictions regarding structure ( docking), dynamics ( molecular dynamics) and, rather limited, about spectra (vibrational Infra Red) can be made. • Due to the low computational effort, force fields are well suited to allow conformational searches. • 4D-QSAR (different docked conformations, e.g. in cytochrome P450) Modern Methods in Drug Discovery WS08/09
Molecular properties from molecular dynamics simulations Binding affinities (actually free energies of binding) DG for ligand binding to enzymes from free energy perturbation calculations Advantage: quite precise predictions Disadvantage: computationally very demanding, thus only feasible for a small number of ligands Lit.: A.R. Leach Molecular Modelling, Longman. Modern Methods in Drug Discovery WS08/09