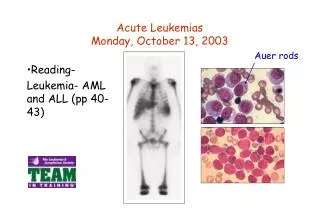
Acute leukemias
Acute leukemias. Кафедра госпитальной терапии Доц. Калинкина Н.В. Acute leukemias. are heterogeneous group of diseases characterized by infiltration of the blood, bone marrow, and other tissues by neoplastic cells (blasts) of the hematopoietic system. . Acute myeloid leukemia.


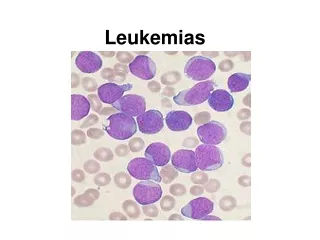
Acute leukemias
E N D
Presentation Transcript
Acute leukemias Кафедра госпитальной терапии Доц. Калинкина Н.В.
Acute leukemias • are heterogeneous group of diseases characterized by infiltration of the blood, bone marrow, and other tissues by neoplastic cells (blasts) of the hematopoietic system.
Acute myeloid leukemia • The incidence of acute myeloid leukemia (AML) is about 3,6 per 100,000 people per year, and the age adjusted incidence is higher in men than in women (1,7-4,4 versus 1,4-3). AML incidence increases with age; it is 1,7 in individuals <65 years and 16.2 in those.65 years. A significant increase in AML incidence has occurred over the past 10 years.
Risk Factors Heredity • Down syndrome • Klinefelter syndrome • congenital neutropenia • Fanconi anemia • neurofibromatosis
Risk Factors Radiation Chemical and other exposures • benzene • smoking • petroleum products • paint • embalming fluids • ethylene oxide • herbicides and pesticides
Risk Factors Drugs • alkylating agent • topoisomerase II inhibitor (etoposid) • chloramphenicol • phenylbutazone • chloroquine • methoxypsoralen
Risk Factors Antecedent haematological disorders • myelodisplastic syndrome • aplastic anaemia • myelofibrosis • paroxismal nocturnal hemoglobinuria • polycithemia rubra vera Familial syndromes
Pathophysiology The underlying pathophysiology consists of a maturational arrest of bone marrow cells in the earliest stages of development. The mechanism of this arrest is under study, but in many cases, it involves the activation of abnormal genes through chromosomal translocations and other genetic abnormalities.
Pathophysiology This developmental arrest results in 2 disease processes. First, a marked decrease in the production of normal blood cells occurs, resulting in varying degrees of anemia, thrombocytopenia, and neutropenia. Second, the rapid proliferation of these cells, along with a reduction in their ability to undergo programmed cell death (apoptosis), results in their accumulation in various organs, most commonly the spleen and liver.
Diagnosis The diagnosis of AML is established by the presence of more than 20% myeloblasts in blood and/or bone marrow according to the World Health Organization classification.
Classification Morphologic
Classification WHO classification
Classification Cytochemical Immunophenotypic Chromosomal • AML with recurrent genetic abnormalities. • AML with t(8;21)(q22;q22) has been associated with M2 • AML with abnormal bone marrow eosinphils (inv(16)(p13q22) or t(16;16)(p13,q22) • Acute promyelocytic leukemia with t(15;17)(q22;q12) • AML with 11q23 abnormalities has been associated with M5 Molecular
Clinical presentation • Anemic syndrome • Infection diseases • Bleeding • Anorexia and weight loss
Clinical presentation • Organ infiltration with leukemic cells • hepatosplenomegaly • lymphadenopathy • gum hypertrophy • leukemia cutis • infiltration of CNS (headache, papilledema, retinal infiltrates, nonfocal neurologic or cranial nerve abnormalities) • Leukostasis (respiratory distress and altered mental status, bone pain )
Lab Studies 1. CBC count with differential demonstrates anemia and thrombocytopenia to varying degrees. Patients with AML can have high, normal, or low WBC counts.
Lab Studies 2. Bone marrow aspiration: The disease then can be classified on the basis of cytochemical stains into any of 7 subtypes, M1-M7. Slides should be stained with myeloperoxidase (or Sudan black), terminal deoxynucleotidyl transferase (TdT) (unless performed by another method, such as flow cytometry), and double esterase to determine the French American British (FAB) type of the leukemia.
Lab Studies AML is defined by greater than 20% blasts in bone marrow. Blasts in bone marrow Normal bone marrow
Lab Studies Сytochemical investigation myeloperoxidase alpha-naphthyl butyrate esterase and chloroacetate esterase stains
Lab Studies Flow cytometry
Lab Studies • Cytogenetic studies • Raise lactic dehydrogenase levels (LDL) • Raise uric acid • An elevated prothrombin time, a decreasing fibrinogen level, the presence of fibrin split products • Liver and renal function must be checked before initiating chemotherapy • Reduce potassium, calcium and magnesium (M4, M5) • If fever is present appropriate steps should be taken to identify and treat infection • Lumbar puncture for those with symptoms of CNS involvment
Clinical remission • The blood neutrophil count must be more then 1500 in microl • The platelet count more then 100000 in microl. • Circulating blasts should be absent. • Bone marrow cellularity should be more then 20% with trilineage maturation. • The bone marrow should contain less then 5% blasts. • Extramedullary leukemia should not be present.
Prognostic factors • Age at diagnosis Chronic and intercurrent diseases impair tolerance to rigorous therapy. • Performance status. • Chromosome finding: t(8;21), inv (16), or t(15,17) - good prognosis; a coplex karyotype, inv (3) or (7) - a very poor prognosis. • Molecular markers. • A prolonged symptomatic interval (more than 1 month before diagnosis of AML) with cytitopenias preceding diagnosis.
Prognostic factors • A history of an antecedent hemotologic disorder. • Other pretreatment clinical featueres that are associated with a lower rate and shorter survival time. • A high presenting leukocyte count. • The FAB classification diagnosis. • Several treatment factors: including achievment of CR, rapidity with which the blast cells disappear from the blood after induction therapy.
Treatment Induction chemotherapy Cytarabine is a cell cycle S-phase specific antimetabolite that becomes phosphorylated intracellularly to an active triphosphate form that interferes with DNA synthesis. Anthracyclines are DNA intercalaters. Their primary mode of action is thought to be inhibition of topoisomerase II, leading to DNA breaks.
Supportive Care • Growth factors • Multilumen right arterial catheters • Adequate and prompt blood bank support: platelet transfusion, red blood cell transfusions • Prophylactic administration of antibiotics in the absence of fever is controversial • Oral nystatin or clotrimazole • Acyclovir prophylaxis • Early initiation of empirical broad-spectrum antibacterial and antifungal antibiotics has significantly reduced the number of patients dying of infectious complications.
Treatment of Promyelocitic Leukemia • Tretinoin is an oral drug that induces the differentiation of leukemic cells bearing the t(15;17); it is not effective in other forms of AML. Trerinoin plus concurrent anthracyline chemotherapy appears to be the safest and most effective treatment for APL.
Postremission Consolidation Therapy • For younger patients, most studies include intensive chemotherapy and allogenic or autologous SCT. High-dose cytarabine is more effective than standard-dose cytarabine. • For older patients, exploration of attenuated intensive therapy that includes either chemotherapy or nonmyeloablative allogeneic SCT has been pursued. In addition, early introduction of new agents is often pursued.
Acute lymphoblastic leukemia • is a malignant (clonal) disease of the bone marrow in which early lymphoid precursors proliferate and replace the normal hematopoietic cells of the marrow.
Pathophysiology • The malignant cells of ALL are lymphoid precursor cells (ie, lymphoblasts) that are arrested in an early stage of development. This arrest is caused by an abnormal expression of genes, often as a result of chromosomal translocations. The lymphoblasts replace the normal marrow elements, resulting in a marked decrease in the production of normal blood cells. The lymphoblasts also proliferate in organs other than the marrow, particularly the liver, spleen, and lymph nodes.
FAB classification • L1 - lymphoid malignencies of small uniform blasts (e.g. typical childhood acute lymphoblastic leukemia): small cells with homogeneous chromatin, regular nuclear shape, small or absent nucleolus, and scanty cytoplasm; subtype represents 25-30% of adult cases
FAB classification • L2 - lymphoid malignencies of uiniform cells with larger and more variable size cells: large and heterogeneous cells, heterogeneous chromatin, irregular nuclear shape, and nucleolus often large; subtype represents 70% of cases (most common)
FAB classification • L3 - lymphoid malignencies of uniform cells with basophilic and sometimes vacuolated cytoplasm (e.g. typical Burkitt’s lymphoma cells). Subtype represents 1-2% of adult cases
Etiology • Epstein-Barr virus • Higher socioeconimic subgroups • Trisomy 21 (Down’s syndrome) • High-energy radiation • Industrial exposure • Exposure to agricultural chemicals • Smoking • Preexisting myeloproliferative disorder
Clinical Features • Bone pain • Hepatosplenomegaly, lymphadenopathy. • Rashes • Left upper quadrant fullness and early satiety due to splenomegaly. • Symptoms related to a large mediastinal mass, such as shortness of breath. • Symptoms of leukostasis (eg, respiratory distress, altered mental status) • Anemic syndrome • Increase risk of infection • Fever • Disseminated intravascular coagulation (hemorrhagic or thrombotic complications)
Lab Studies • A CBC count: anemia,thrombocytopenia, a high, normal, or low WBC count, neutropenia, blasts
Lab Studies • Bone marrow aspiration and biopsy
Lab Studies A negative peroxidase stain AML ALL
Lab Studies • Flow cytometry: CD3 (T-lineage ALL) CD19 (B-lineage ALL) • Cytogenetics: t(22;9) t(4;11) t(2;8) t(8;14))
Lab Studies • Coagulogramm: elevated prothrombin time decreased fibrinogen levels presence of fibrin split products • Chemistry profile: elevated lactic dehydrogenase level elevated uric acid level liver function tests BUN/creatinine determinations
Lab Studies • Blood cultures • Imaging studies • ECG • Histologic findings
Prognosis Genetic abnormalities: t(9;22) - poor outlook t(4;11) - younger age, female predominance, high white cell counts, and L1 morphology t(8;14) - older age, male predominance, frequent CNS involvement, and L3 morphology Both are associated with a poor prognosis.
Medical Care • Induction therapy: 4-drug regimen of vincristine, prednisone, anthracycline, and cyclophosphamide or L-asparaginase or a 5-drug regimen of vincristine, prednisone, anthracycline, cyclophosphamide, and L-asparaginase given over the course of 4-6 weeks. • Consolidation therapy: a standard 4- to 5-drug induction usually include consolidation therapy with Ara-C in combination with an anthracycline or epipodophyllotoxin. • Maintenance • CNS prophylaxis
Supportive Care • Replacement of blood products: packed red blood cells, platelets, fresh frozen plasma • Antibiotics: a third-generation cephalosporin (or equivalent) with an aminoglycoside. Patients with persistent fever after 3-5 days of antibacterial antibiotics have amphotericin added to their regimen. • The use of prophylactic antibiotics in neutropenic patients who are not febrile is controversial. A commonly used regimen includes ciprofloxacin (500 mg orally twice daily, fluconazole (Diflucan) (200 mg orally daily), and acyclovir (200 mg orally 5 times/d).
Supportive Care • Growth factors • Allopurinol 300 mg 1-3 times/d • Central venous catheter
Prognosis • Good risk includes (1) no adverse cytogenetics, (2) age younger than 30 years, (3) WBC count of less than 30,000/mL, and (4) complete remission within 4 weeks. • Intermediate risk does not meet the criteria for either good risk or poor risk. • Poor risk includes (1) adverse cytogenetics [(t9;22), (4;11)], (2) age older than 60 years, (3) precursor B-cell WBCs with WBC count greater than 100,000/mL, or (4) failure to achieve complete remission within 4 weeks.