Download

1 / 47
490 likes | 846 Views
AGENESIA DEL CUERPO CALLOSO QUISTE INTERHEMISFÉRICO. Dr. José Miguel Cárdenas Galli Residente Neurología Infantil HCSBA. Dra. Susana Lara Mora Neuróloga Infantil HCSBA.

E N D
AGENESIA DEL CUERPO CALLOSO QUISTE INTERHEMISFÉRICO Dr. José Miguel Cárdenas Galli Residente Neurología Infantil HCSBA Dra. Susana Lara Mora Neuróloga Infantil HCSBA
La agenesia del cuerpo calloso se define como el defecto primario total o parcial de las fibras comisurales que cruzan la línea media y conectan los dos hemisferios cerebrales. INTRODUCCIÓN MARÍA EUGENIA HÜBNER GUZMÁN. Malformaciones congénitas: diagnóstico y manejo neonatal. Editorial Univerditaria. 2005
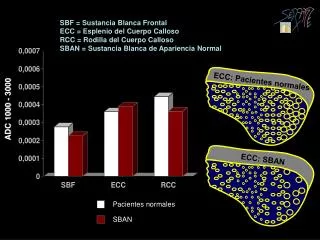
ANATOMÍA • Es la comisura interhemisférica de mayor tamaño y conecta transversalmente ambos hemisferios. • Su morfología es rectangular y de mayores dimensiones en dirección rostro-caudal que dorso-ventral. • Su tamaño es aproximadamente de 10cms. en sentido anteroposterior, 4cms. en su parte más anterior frontal y 6cms. en la parte más posterior, cercana a la parte occipital • Está formado aproximadamente por unos 180-200 millones de axones. • Es capaz de transportar unos 4.000 millones de impulsos por segundo. QUINTERO-GALLEGO, MANAUT, RODRÍGUEZ, ET AL. Desarrollo diferencial del cuerpo calloso en relación con el hemisferio cerebral. Revista Española de Neuropsicología 5, 1:49-64 (2003)
ANATOMÍA Hemisferio derecho, (1) Rodilla del cuerpo calloso, (2) Esplenio del cuerpo calloso, (3) Tronco del cuerpo calloso, (4) Polo anterior y (5) Polo posterior. OLAVE, E.; TORREZ, J. C.; RIQUELME, N. ; IBACACHE, L. & BINVIGNAT, O. Características biométricas del cuerpo calloso en individuos chilenos. Int. J. Morphol., 30(4):1449-1452, 2012.
EMBRIOLOGÍA ELIZABETH EMMA PALMER, DAVID MOWAT. Agenesis of the Corpus Callosum: A ClinicalApproachto Diagnosis. American Journal of MedicalGeneticsPart C (Seminars in MedicalGenetics) 166C:184–197 (2014) La agenesia del cuerpo calloso se producirá por una agresión sobre la la lámina terminal o la masa comisural (7 a 12 semanas de gestación). MARÍA EUGENIA HÜBNER GUZMÁN. Malformaciones congénitas: diagnóstico y manejo neonatal. Editorial Univerditaria. 2005
EMBRIOLOGÍA T. W. SADLER, JAN LANGMAN. Langman embriología médica: con orientación clínica. Editorial Médica Panamericana. 2007
La real incidencia, prevalencia y mortalidad del cuerpo calloso resulta muy difícil de establecer. • La incidencia de agenesia del cuerpo calloso va de 0,7% a 5,3% población general a 2% a 3% en niños con RDSM. • La prevalencia de agenesia del cuerpo calloso va de 0,5 por 10,000 en población general a 230 – 600 por 10,000 en niños con RDSM. • En todas las series es más frecuente en hombres que en mujeres con una proporción 3:2. EPIDEMIOLOGÍA MARÍA EUGENIA HÜBNER GUZMÁN. Malformaciones congénitas: diagnóstico y manejo neonatal. Editorial Univerditaria. 2005 ELIZABETH EMMA PALMER, DAVID MOWAT. Agenesis of the Corpus Callosum: A ClinicalApproachto Diagnosis. American Journal of MedicalGeneticsPart C (Seminars in MedicalGenetics) 166C:184–197 (2014)
ETIOLOGÍA ELIZABETH EMMA PALMER, DAVID MOWAT. Agenesis of the Corpus Callosum: A ClinicalApproachto Diagnosis. American Journal of MedicalGeneticsPart C (Seminars in MedicalGenetics) 166C:184–197 (2014)
ETIOLOGÍA ELIZABETH EMMA PALMER, DAVID MOWAT. Agenesis of the Corpus Callosum: A ClinicalApproachto Diagnosis. American Journal of MedicalGeneticsPart C (Seminars in MedicalGenetics) 166C:184–197 (2014)
DIAGNÓSTICO • El diagnóstico prenatal se efectúa mediante ecografía y RM y permite realizar el diagnóstico a partir de las 20 semanas de gestación. • Eldiagnósticoposnatal se efectúa medianteecografía, TC o RM. T. GONÇALVES-FERREIRA, ET AL. Agenesia del cuerpo calloso. REV NEUROL 2003; 36 (8): 701-706
DIAGNÓSTICO En el corte transversal se debe buscar la indemnidad del cavumseptumpellucidum. En la ACC éste estará ausente o tendrá una configuración anómala. Además evaluar los ventrículos laterales buscando anomalías: cuernos anteriores lateralizados o cuernos posteriores en lágrima. ELIZABETH EMMA PALMER, DAVID MOWAT. Agenesis of the Corpus Callosum: A ClinicalApproachto Diagnosis. American Journal of MedicalGeneticsPart C (Seminars in MedicalGenetics) 166C:184–197 (2014)
DIAGNÓSTICO En el corte coronal se debe buscar la indemnidad del cavumseptumpellucidum. En la ACC éste estará ausente o tendrá una configuración anómala. Además evaluar los ventrículos laterales buscando anomalías: cuernos anteriores lateralizados con patrón en "cuerno de vaca" ELIZABETH EMMA PALMER, DAVID MOWAT. Agenesis of the Corpus Callosum: A ClinicalApproachto Diagnosis. American Journal of MedicalGeneticsPart C (Seminars in MedicalGenetics) 166C:184–197 (2014)
DIAGNÓSTICO En el corte sagital se debe buscar anormalidades en el patrón de giros de la corteza con una disposición radiada. ELIZABETH EMMA PALMER, DAVID MOWAT. Agenesis of the Corpus Callosum: A ClinicalApproachto Diagnosis. American Journal of MedicalGeneticsPart C (Seminars in MedicalGenetics) 166C:184–197 (2014)
DIAGNÓSTICO En el corte coronal y sagital buscar los haces de Probst que son fascículos de fibras nerviosas que bordean los ventrículos laterales y que proceden de las fibras comisurales que no cruzaron. T. GONÇALVES-FERREIRA, ET AL. Agenesia del cuerpo calloso. REV NEUROL 2003; 36 (8): 701-706
Laagenesia del cuerpo calloso se asocia hasta en un 80% de los casos a otras malformaciones encefálicas. • Quisteinterhemisférico. • Alteraciones de la migración neuronal. • Holoprosencefalia. • Malformación de Dandy-Walker. • Malformación de Chiari II. • Quiste aracnoideo. MALFORMACIONES ASOCIADAS T. GONÇALVES-FERREIRA, ET AL. Agenesia del cuerpo calloso. REV NEUROL 2003; 36 (8): 701-706
AGENESIA CUERPO CALLOSO + QUISTEINTERHEMISFÉRICO
AGENESIA CUERPO CALLOSO + QUISTEINTERHEMISFÉRICO JAMESBARKOVICH, MD; ERIN M. SIMON, MD;CHRISTOPHER A. WALSH, MD, PHD. Callosalagenesiswithcyst.A betterunderstandingandnewclassification. NEUROLOGY 2001;56;220-227.
AGENESIA CUERPO CALLOSO + QUISTEINTERHEMISFÉRICO JAMESBARKOVICH, MD; ERIN M. SIMON, MD;CHRISTOPHER A. WALSH, MD, PHD. Callosalagenesiswithcyst.A betterunderstandingandnewclassification. NEUROLOGY 2001;56;220-227.
AGENESIA CUERPO CALLOSO + QUISTEINTERHEMISFÉRICO JAMESBARKOVICH, MD; ERIN M. SIMON, MD;CHRISTOPHER A. WALSH, MD, PHD. Callosalagenesiswithcyst.A betterunderstandingandnewclassification. NEUROLOGY 2001;56;220-227.
Posiblemente, las mismas moléculas señalizadoras (integrinas, semaforinas, netrinas) que guían los axonescomisurales a sus respectivos puntos de cruce en la línea media, también guiarían la migración de las neuronas desde la zona germinal hacia la corteza en desarrollo. AGENESIA CUERPO CALLOSO + QUISTEINTERHEMISFÉRICO JAMESBARKOVICH, MD; ERIN M. SIMON, MD;CHRISTOPHER A. WALSH, MD, PHD. Callosalagenesiswithcyst.A betterunderstandingandnewclassification. NEUROLOGY 2001;56;220-227.
SÍNDROME DE AICARDI Dr. José Miguel Cárdenas Galli Residente Neurología Infantil HCSBA Dra. Susana Lara Mora Neuróloga Infantil HCSBA
INTRODUCCIÓN J. AICARDI.AicardiSyndrome.BRAIN & DEVELOPMENT 27 (2005) 164–171.
La epidemiologíaes poco conocida por tratarse de una enfermedad muy rara. • Laprevalencia reportada es de 300 a 500 casos publicados a la fecha y la prevalencia estimadaes de 2-15/100.000 niñas y en unos 4.000 pacientes en el mundo. • Laincidenciase sitúa entre 1 por 93.000 en Europa y 1 por 167.000 casos en EEUU. • En series de espasmos infantiles, el SA se ha considerado la etiología responsable de la epilepsia entre 0,5%y el 4% de los casos. EPIDEMIOLOGÍA KRONER ET AL. Incidence, Prevalence, andSurvival of AicardiSyndrome. JOURNAL OF CHILD NEUROLOGYVolume 23 Number 5 May 2008 531-535
El síndrome de Aicardies una enfermedad de presumible origen genético. • Se presume que se debe a mutaciones dominantes de novo en un gen ligado al cromosoma X. • Su presentación ocasional en varones con síndrome de Klinefelter (47,XXY) apoya hipótesis de un trastorno de herencia dominante ligada al sexo, letal en las etapas tempranas de la gestación para los embriones XY. • Sólo presentación excepcional en dos hermanas apoya hipótesis de mutaciones de novo. ETIOLOGÍA J. AICARDI.AicardiSyndrome.BRAIN & DEVELOPMENT 27 (2005) 164–171.
Es uno de los elementos diagnósticos que definen el síndrome de Aicardi. • Suelen aparecer de manera precoz, edad media de presentación entre el 3 y 4 mes de vida. • Suelen ser asimétricos, comprometiendo en mayor medida a un hemicuerpo. • EEG interictal característico con patrón estallido/supresión unilaterales o bilaterales asincrónicos. • EEG ictal con ondas lentas de alto voltaje antecedidas por actividad rítmica de bajo voltaje que se superimpone en el inicio de las ondas lentas. • Tanto los espasmos como el trazado suelen presentar escasas modificaciones en el tiempo. ESPASMOS INFANTILES J. AICARDI.AicardiSyndrome.BRAIN & DEVELOPMENT 27 (2005) 164–171.
La agenesia del cuerpo calloso es el elemento central en la esfera neuroanatómica • Laagenesia del cuerpo calloso es total en la mayoría de los casos aunque están descritas las formas parciales. • Inicialmente descrita como elemento único. Con los avances en neuroimagen esto cambió. • Otras alteraciones neuroanatómicas descritas: displasiacortical, heterotopias, formaciones quísticas, anomalías vermianas, papilomas del plexo coroídeo, etc. ALTERACIONES NEUROLÓGICAS J. AICARDI.AicardiSyndrome.BRAIN & DEVELOPMENT 27 (2005) 164–171.
Principal elemento diagnóstico por su carácter patognomónico. • Las alteraciones ocularestienen una importancia considerable en el diagnóstico del síndrome de Aicardi. • Las lagunas coriorretinales son consideradas patognomónicas de la enfermedad. • Otras alteraciones oftalmológicas:coloboma del disco óptico, microftalmia, depósitos pigmentariosretinales, etc. ALTERACIONES OFTÁLMICAS J. AICARDI.AicardiSyndrome.BRAIN & DEVELOPMENT 27 (2005) 164–171.
ALTERACIONES OFTÁLMICAS J. AICARDI.AicardiSyndrome.BRAIN & DEVELOPMENT 27 (2005) 164–171.
Anormalidades Costales • Ausencia de costillas unilateral o bilateral. • Bifurcaciones costales unilateral o bilateral. OTRAS ALTERACIONES Anormalidades Vertebrales • Escoliosis. • Vértebra en mariposa. • Hemivértebra J. AICARDI.AicardiSyndrome.BRAIN & DEVELOPMENT 27 (2005) 164–171.
DISMORFIAS FACIALES Maxilar prominente. Puente nasal bajo. V. REIDSUTTON ET AL. Facial andPhysicalFeatures of AicardiSyndrome. AMERICAN JOURNAL OF MEDICALGENETICS 138A:254–258 (2005)
DISMORFIAS FACIALES NarinasEvertidas. Cejas laterales despobladas. V. REIDSUTTON ET AL. Facial andPhysicalFeatures of AicardiSyndrome. AMERICAN JOURNAL OF MEDICALGENETICS 138A:254–258 (2005)
Puesto que aún no se ha identificado un gen ni un locus asociado a este síndrome, el diagnóstico continúa basándose en la asociación de síntomas y signos característicos. DIAGNÓSTICO J.A. FERNÁNDEZ-RAMOS, ET AL. Síndrome de Aicardi: estudio retrospectivo de una serie de siete casos. REVNEUROL 2013; 57 (11): 481-488
Manejo con enfoque multidisciplinario orientado a las complicaciones a largo plazo • Los espasmos suelen ser refractarios a FAE y el manejo con ACTH no estaría indicado por su baja efectividad y riesgos asociados. No existen datos suficientes en relación a resección quirúrgica y estimulación vagal. • El manejo ortopédico es importante para evitar escoliosis y contracturas. • El manejo de los cuadros infecciosos debe ser particularmente agresivo. MANEJO J. AICARDI.AicardiSyndrome.BRAIN & DEVELOPMENT 27 (2005) 164–171.
El pronóstico es grave, lo habitual es que curse con epilepsia refractaria y una discapacidad intelectual severa. • Menos del 5% de los casos llega a desarrollar algún lenguaje expresivo. • Menos del 25% de los casos adquiere la marcha autónoma. PRONÓSTICO J. AICARDI.AicardiSyndrome.BRAIN & DEVELOPMENT 27 (2005) 164–171.
La mortalidad es elevada en la infancia con una tasa de sobrevida del 76% a los 6 años y del 40% a los 14 años. La principal causa son las infecciones pulmonares. PRONÓSTICO KRONER ET AL. Incidence, Prevalence, andSurvival of AicardiSyndrome. JOURNAL OF CHILD NEUROLOGYVolume 23 Number 5 May 2008 531-535
SÍNDROME DE DELLEMAN ÓCULO-CEREBRO-CUTÁNEO Dr. José Miguel Cárdenas Galli Residente Neurología Infantil HCSBA Dra. Susana Lara Mora Neuróloga Infantil HCSBA
INTRODUCCIÓN U MOOG, M C JONES, L M BIRD, W B DOBYNS. Oculocerebrocutaneoussyndrome: the brainmalformation defines a core phenotype. J MEDGENET 2005;42:913–921.
Quiste orbitario asociado a apéndices cutáneos periorbitarios • El quiste orbitario es la principal alteración oftalmológica del síndrome de Delleman y forma parte de su triada. • Otras alteraciones oftalmológicas descritas son: microftalmia, anoftalmia, coloboma de los párpados. ALTERACIONES OFTÁLMICAS U MOOG, M C JONES, L M BIRD, W B DOBYNS. Oculocerebrocutaneoussyndrome: the brainmalformation defines a core phenotype. J MEDGENET 2005;42:913–921.
ALTERACIONES OFTÁLMICAS Quiste orbitario. Apéndices cutáneos perioculares. Quiste orbitario. Anoftalmia. VIPUL ARORA, USHA R KIM, HADI M KHAZEI. Dellemansyndrome: ‘Oculocerebrocutaneoussyndrome'. INDIAN J OPHTHALMOL: 2009;57:387-389
Focos de ausencia de tejido dérmico/epidérmico o alteración en su formación • Focos de aplasia cutis o hipoplasia cutis en forma de placa o perforaciones. • Otras alteraciones dermatológicas incluyen los apéndices cutáneos periorbitarios y la alopecia. ALTERACIONES CUTÁNEAS U MOOG, M C JONES, L M BIRD, W B DOBYNS. Oculocerebrocutaneoussyndrome: the brainmalformation defines a core phenotype. J MEDGENET 2005;42:913–921.
Las malformaciones del SNC presentan unfenotipo ampliado • Los hemisferios muestran superficie irregular, sulcación reducida, con aumento del grosor y disminución de la sustancia blanca. • Los ventrículos laterales suelen estar aumentados de tamaño y el cuarto ventrículo se comunica con la fosa posterior. • El cuerpo calloso puede estar ausente o alterado en su morfología con o sin presencia de quistes interhemisféricos. • La fosa posterior puede presentar múltiples alteraciones: quistes porencefálicos, agenesia del vermiscerebeloso, etc. ALTERACIONES NEUROLÓGICAS U MOOG, M C JONES, L M BIRD, W B DOBYNS. Oculocerebrocutaneoussyndrome: the brainmalformation defines a core phenotype. J MEDGENET 2005;42:913–921.
¿ORIGEN GENÉTICO? PATOGENIA Alteraciones de estructuras romboencefálicas. Preponderancia de casos de sexo masculino. ¿FALLA EN ROMBÓMERO 1? ¿HERENCIA LIGADA A X? U MOOG, M C JONES, L M BIRD, W B DOBYNS. Oculocerebrocutaneoussyndrome: the brainmalformation defines a core phenotype. J MEDGENET 2005;42:913–921.
ANÁLISIS DEL CASO Dr. José Miguel Cárdenas Galli Residente Neurología Infantil HCSBA Dra. Susana Lara Mora Neuróloga Infantil HCSBA
QUINTERO-GALLEGO, MANAUT, RODRÍGUEZ, ET AL. Desarrollo diferencial del cuerpo calloso en relación con el hemisferio cerebral. Revista Española de Neuropsicología 5, 1:49-64 (2003). • MARÍA EUGENIA HÜBNER GUZMÁN. Malformaciones congénitas: diagnóstico y manejo neonatal. Editorial Univerditaria. 2005 . • OLAVE, E.; TORREZ, J. C.; RIQUELME, N. ; IBACACHE, L. & BINVIGNAT, O. Características biométricas del cuerpo calloso en individuos chilenos. Int. J. Morphol., 30(4):1449-1452, 2012. • ELIZABETH EMMA PALMER, DAVID MOWAT. Agenesis of the Corpus Callosum: A ClinicalApproachto Diagnosis. American Journal of MedicalGeneticsPart C (Seminars in MedicalGenetics) 166C:184–197 (2014). • T. W. SADLER, JAN LANGMAN. Langman embriología médica: con orientación clínica. Editorial Médica Panamericana. 2007. • T. GONÇALVES-FERREIRA, ET AL. Agenesia del cuerpo calloso. REV NEUROL 2003; 36 (8): 701-706. • JAMESBARKOVICH, MD; ERIN M. SIMON, MD;CHRISTOPHER A. WALSH, MD, PHD. Callosalagenesiswithcyst.A betterunderstandingandnewclassification. NEUROLOGY 2001;56;220-227. • KRONER ET AL. Incidence, Prevalence, andSurvival of AicardiSyndrome. JOURNAL OF CHILD NEUROLOGYVolume 23 Number 5 May 2008 531-535. • V. REIDSUTTON ET AL. Facial andPhysicalFeatures of AicardiSyndrome. AMERICAN JOURNAL OF MEDICALGENETICS 138A:254–258 (2005). • J. A. FERNÁNDEZ-RAMOS, ET AL. Síndrome de Aicardi: estudio retrospectivo de una serie de siete casos. REVNEUROL 2013; 57 (11): 481-488. • U MOOG, M C JONES, L M BIRD, W B DOBYNS. Oculocerebrocutaneoussyndrome: the brainmalformation defines a core phenotype. J MEDGENET 2005;42:913–921. • VIPUL ARORA, USHA R KIM, HADI M KHAZEI. Dellemansyndrome: ‘Oculocerebrocutaneoussyndrome'. INDIAN J OPHTHALMOL: 2009;57:387-389 BIBLIOGRAFÍA