Download

1 / 21
210 likes | 332 Views
Reviewing the Jukes-Cantor model for estimating evolutionary distances between sequences and building phylogenetic trees based on rate matrices in molecular evolution.

E N D
Molecular evolution, cont.Estimating rate matrices Lecture 15, Statistics 246 March 11, 2004
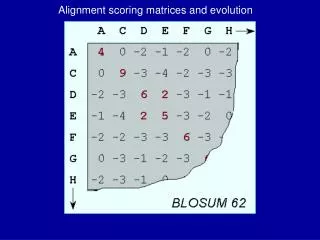
REVIEW:The Jukes-Cantor model (1969) • Q = • P(t) = C o m m o n a n c e s t o r o f h u m a n a n d o r a n g . t t i m e u n i t s h u m a n ( n o w ) • Consider e.g. the 2nd position in a-globin2 Alu1. r = (1+3e4t)/4, s = (1 e4t)/4.
REVIEW:Jukes-Cantor adjustment common ancestor still 2nd position in a-globin Alu 1 Assume that the common ancestor has A, G, C or T with probability 1/4. C human G orang 3/4 Then the chance of the nt differing p≠ = 3/4 (1 e8t) = 3/4 (1 e4k/3), since k =2 3t Solving for k = estimating distance in PAMs t
REVIEW: Estimating the evolutionary distance between two sequences • Suppose two aligned protein sequences a1…anand b1…bn are separated by t PAMs. • Under a reversible substitution model that is i.i.d across sites, the likelihood function of t is • L(t) = pr(a1…an,b1…bn | model) • = k F(t,ak,bk) • = a,b F(t,a,b)c(a,b), • where c(a,b) = # {k : ak = a, bk = b}, and • F(t,a,b) = (a)P(t,a,b) = (b)P(t,b,a) = F(t,b,a). • Maximizing this quantity in t with F known gives the maximum likelihood estimate of t. This generalizes the distance correction with Jukes-Cantor.
Acknowledgement • Von Bing Yap, • for joint work summarized here and in the previous and next lecture.
From aligned DNA or protein sequences to evolutionary trees The starting point for a molecular phylogenetic analysis is a set of sequences, almost always aligned. The end result is almost always a tree. Along the way, attention needs to be paid to substitution process operating in the sequences, and to possible rate variationalong the sequences, and down the tree. The two main approaches to tree building are a) distance-based methods, which work from pairwise distances between the sequences, and b) character-based methods, which work directly from the multiply aligned sequences. We’ll briefly mention both, referring you to the literature for fuller details. Both make use of rate matrices.
Building trees: distance methods There are many ways of building trees using distance methods. All start by computing the pairwise distances between the sequences to be at the tips of the tree, usually along the lines we discussed in the last lecture, i.e. ML distance, using a rate matrix. One of the oldest distance methods, still widely used, though rather discredited in the molecular evolutionary context , is UPGMA. This stands for unweighted pair group method with arithmeticmeans. It is easy to understand quickly, and so I will describe it. I don’t recommend it. A more recent and much more satisfactory method in molecular evolution is the neighbour-joiningapproach,abbrev. NJ. It takes longer to explain, so I won’t give it here. There are many places where the details of this and other methods are given including Durbin et al (1998), and the recent excellent book by the master: Joseph Felsenstein, Inferring phylogenies, Sinauer, 2004.
Beta-globins revisited 10 20 30 40 BG-human M V H L T P E E K S A V T A L W G K V N V D E V G G E A L G R L L V V Y P W T Q BG-macaque - . . . . . . . . N . . . T . . . . . . . . . . . . . . . . . . . . . . . . . . BG-bovine - - M . . A . . . A . . . . F . . . . K . . . . . . . . . . . . . . . . . . . . BG-platypus - . . . S G G . . . . . . N . . . . . . I N . L . . . . . . . . . . . . . . . . BG-chicken . . . W . A . . . Q L I . G . . . . . . . A . C . A . . . A . . . I . . . . . . BG-shark - . . W S E V . L H E I . T T . K S I D K H S L . A K . . A . M F I . . . . . T 50 60 70 80 BG-human R F F E S F G D L S T P D A V M G N P K V K A H G K K V L G A F S D G L A H L D BG-macaque . . . . . . . . . . S . . . . . . . . . . . . . . . . . . . . . . . . . N . . . BG-bovine . . . . . . . . . . . A . . . . N . . . . . . . . . . . . D S . . N . M K . . . BG-platypus . . . . A . . . . . S A G . . . . . . . . . . . . A . . . T S . G . A . K N . . BG-chicken . . . A . . . N . . S . T . I L . . . M . R . . . . . . . T S . G . A V K N . . BG-shark . Y . G N L K E F T A C S Y G - - - - - . . E . A . . . T . . L G V A V T . . G 90 100 110 120 BG-human N L K G T F A T L S E L H C D K L H V D P E N F R L L G N V L V C V L A H H F G BG-macaque . . . . . . . Q . . . . . . . . . . . . . . . . K . . . . . . . . . . . . . . . BG-bovine D . . . . . . A . . . . . . . . . . . . . . . . K . . . . . . . V . . . R N . . BG-platypus D . . . . . . K . . . . . . . . . . . . . . . . N R . . . . . I V . . . R . . S BG-chicken . I . N . . S Q . . . . . . . . . . . . . . . . . . . . D I . I I . . . A . . S BG-shark D V . S Q . T D . . K K . A E E . . . . V . S . K . . A K C F . V E . G I L L K 130 140 BG-human K E F T P P V Q A A Y Q K V V A G V A N A L A H K Y H . means same as reference sequence - means deletion BG-macaque . . . . . Q . . . . . . . . . . . . . . . . . . . . . BG-bovine . . . . . V L . . D F . . . . . . . . . . . . . R . . BG-platypus . D . S . E . . . . W . . L . S . . . H . . G . . . . BG-chicken . D . . . E C . . . W . . L . R V . . H . . . R . . . BG-shark D K . A . Q T . . I W E . Y F G V . V D . I S K E . .
UPGMA tree for beta-globins BG-shark BG-chicken BG-platypus BG-bovine BG-macaque BG-human 1
Neighbor-joining tree for globins myo-human alpha-human epsilon-human gamma-human beta-human delta-human
Today’s main task • We will discuss three methods of estimating a calibratedreversiblerate matrixQ, given aligned leaf sequences on multiple unrooted phylogenetic trees whose topologies and branch lengths are known. All three methods are consistent, in the sense that the methods are asymptotically unbiased as the sequences become infinitely long. Moreover, evolutionary distances between sequences are explicitly accounted for, unlike with the PAM method. • The maximum likelihood (ML) method is natural for any parametric model, and has well-known theoretical properties. Maximum partial likelihood (MPL) is particularly well-suited to Markov processes, and can be efficiently implemented via an EM algorithm. The resolvent method (RES) is quite a different technique. • In practice, the phylogenetic tree can also be estimated from the data. When the tree topology is known, e.g. when there are just 2 or 3 leaf nodes, the branch lengths can be estimated by ML, given the rate matrix. One can estimate both the rate matrix and the branch lengths by alternating between the two steps: estimate Q given the branch lengths; estimate the branch lengths given Q. Estimating the tree topology as well is a harder problem, on which much has been written.
Parametrization of reversible rate matrices • A transition matrix P(t) with stationary distribution is reversible if for all a,b • (a)P(a, b;t) = (b)P(b,a;t). • If P(t) = exp tQ, then we can write P(t) = I + tQ for small t, and conclude that if P is reversible, then for all a,b we have • (a)Q(a, b) = (b)Q(b,a). (*) • Since (a)>0 for all a may be readily assumed, write R(a,b) = (a)-1Q(b,a). Then (*) implies that R is symmetric (check), and so if we write for the diagonal matrix with the value (a) in row-column a, we have proved that for a reversible chain, Q = R for a unique symmetric matrix R. • Now let us see that such Q are diagonalizable. Let A be the square root of . Then Q = R = A-1(ARA)A, where ARA is symmetric. We can now write ARA= VV’, where V is orthogonal and diagonal. Then Q is seen to be diagonalizable: Q = A-1VV’A. This makes lots of things easier when we deal with numerical work involving the class REV of reversible rate matrices.
Maximum likelihood • Suppose that the ancestral sequences at the internal nodes are known. Take any leaf sequence for the root, cf last lecture, let ti be the length of the ith branch, and let (i) be the frequency table for the pair of sequences connected by this branch: • (i, a, b) = |{ancestor =a, descendant = b}|. • Denote the row sum of the frequency table corresponding to the root node by f. Then the probability of all sequences is • a(a)f(a) ia,bP(ti ,a,b)(i, a, b) . • By reversibility, this value is independent of the choice of root. • In almost all applications, the ancestral sequences are unknown. Imputation by parsimony is biased when the sequences are not closely related, and even when they are, it is better to weight the unobserved sequences according to some probabilistic model. In principle we can sum the above probability over all possible ancestral sequences to get the probability of the observed sequences. Felsenstein has given an efficient algorithm for doing this, by recursively moving up the nodes to the root. It is a tree version of one of the HMM calculations.
Maximum likelihood, cont. • In order to maximize the likelihood, we need to choose a parametrization. A natural choice is to use the equilibrium frequencies, and the top off-diagonal elements of a symmetric matrix R in the factorization Q = R, with constraints • for all a, (a) ≥ 0, • for all a<b, R(a,b) ≥ 0, • a(a) = 1, • 2a<b (a)(b)R(a,b) = 0.01. • The last constraint is for calibration. Even for the simplest case, that is where each tree consists of a pair of sequences, it is difficult to get closed-form expressions for the MLE, and so numerical maximization must be used.
Maximum likelihood, cont. • If the tree consists of just two sequences aand b, the likelihood function depends on the tree only through the evolutionary distancet between the sequences, and can be written out explicitly. Let denote the frequency table for the pair, see two slides back. • Then the likelihood has the multinomial form we saw for estimating the distance between two sequences, namely • a,bF(t,a,b)(a,b) • where the matrix F(t) represents the joint distribution of homologous residues at distance t, given by • F(t,a,b) = (a)P(t,a,b) = (b)P(t,b,a) = F(t,b,a). • The symmetry of F(t) is a consequence of reversibility, and embodies the fact that any intermediate sequence between a and b may be regarded as the ancestor.
Maximum partial likelihood • For a given tree, instead of maximizing the probability of the leaf sequences, one can maximize their conditional probability given the ancestral sequence. This yields the maximum partial likelihood estimator (MPLE), which can be proved consistent, though less efficient, in comparison with the MLE. If all ancestral sequences are observed, and all branch lengths have the same lengtht, then the partial likelihood is • ia,bP(t,a,b)(i, a, b) • = a,bP(t,a,b)C(a, b), where C(a,b) =i (i,a,b). • The MPLE for P(t) is readily seen to be • P^(t,a,b) = C(a,b) / bC(a,b), • which is just Dayhoff’s P. Of course the assumptions in blue are unrealistic.
Maximum partial likelihood, cont. • When the ancestral sequences are unknown, we may regard any leaf as the ancestor, since the substitution process is reversible. Fix a leaf sequence, and consider the relabelled tree with that leaf as root. Then, summing over all possible intermediate sequences (internal nodes) yields the partial likelihood based on the observed sequences. As with ML, numerical maximization is necessary, but in this case, the partial likelihood can be efficiently maximized using an EM algorithm, as shown by Holmes and Rubin (2002). • We present the EM algorithm for two sequences generated by a discrete time Markov chain. The extension to continuous time is straightforward. If time permits, we’ll discuss the tree (multiple sequence) version. The EM algorithm, unlike the ML, does not impose reversibility on its estimates. It works on any time homogeneous substitution process on a rooted tree, provided the root sequence is observed. Further, the initial distribution need not be the stationary distribution. In practice, the root sequence is almost never observed, so by assuming reversibility, the EM can be applied starting from any leaf. To ensure that the EM leads to a reversible rate matrix, the frequency tables are symmetrized at the start.
Pair EM, discrete-time case. • Let P be a reversible transition matrix, with equilibrium and initial distribution . Let the sequence at time s be Xs,1, ….,Xs,n. Let t be a fixed position integer, and suppose that only sequences of length n at times 0 and t are observed. We denote the observed data by O, and the fulldata by F. For s=1,…t, let (s) be the frequency table for transitions between s-1 and s: • (s,a,b) = |{k: Xs-1,k = a, Xs,k = b}|. • Then the partial likelihood for the full data is
PMLE, cont. • The key idea of the EM algorithm is that replacing the unobserved counts by their conditional expectations given the observed data, evaluated at the current estimate of P, gives a new estimate with a higher partial likelihood. The algorithm iterates between two steps: • E-step: given a current estimate P0, compute the quantity • G(P1,P0) = E0,P0 {log L(P1, F | O)} • M-step: maximize G(P1,P0) as a function of P1 . • By site independence, G(P1,P0) is a sum of similar terms. We fix a site, and suppose that X0 = a, Xt = b. Its contribution to G(P1,P0) is then
PMLE, completed. The high powers of P are easily computed when Q, and hence P is diagonalizable. If there are independent sequences separated by possibly different known times, then in the E-step, G( P0, P1) is just the sum of the contributions from the pairs.