Pharmacokinetics
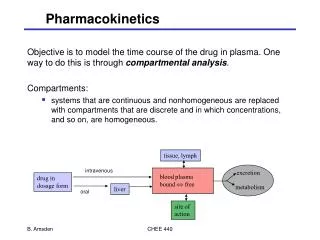
Pharmacokinetics. Pharmacokinetics is the study of those rate processes involved in the absorption, distribution , metabolism, and excretion of drugs (drug disposition) and their relationship to the pharmacological, therapeutic or toxic response in animals and man.


Pharmacokinetics
E N D
Presentation Transcript
Pharmacokinetics Pharmacokinetics is the study of those rate processes involved in the absorption, distribution, metabolism, and excretion of drugs (drug disposition) and their relationship to the pharmacological, therapeutic or toxic response in animals and man. Metabolism + Excretion = Elimination
Absorption - Distribution-Metabolism-Excretion ABSORPTION DISTRIBUTION i.v. DISTRIBUTION LIVER: M + BE KIDNEY: Ur. E
Pharmacokinetics techniques • Pharmacokinetic techniques attempt to mathematically define the time course for drug in the body by assaying for drug and metabolites in readilly accesible fluids such as blood and urine.
Pharmacokinetics techniques • The goal is to quantitatively account for the amount of drug which has entered the body (bioavailable dose) and to estimate the rate by which it is cleared from the body.
Pharmacokinetics techniques • The mathematical descriptions use PK variables (PK parameters) for modelling of the time-course of a drug in plasma or other fluids (concentration versus time curves). PK parameters are than used to calculate the rate of dosing.
Pharmacokinetic parameters • apparent volume of distribution Vd • total clearance Cl • elimination half-life t1/2 • bioavailability F
Distribution Drug distribution means the reversible transfer of drug from one location to another within the body. Once a drug has entered the vascular system it becomes distributed throughout the various tissues and body fluids in a pattern that reflects the physico-chemical nature of the drug and the ease with which it penetrates different membranes.
Factors affecting drug distribution: • Rate of distribution - Membrane permeability - Blood perfusion of organs and tissues • Extent of Distribution - Lipid Solubility - pH - pKa (pH-partition theory for ionizable molecules) - Plasma protein binding - Intracellular binding
Plasma protein binding: • Extensive plasma protein binding will cause more drug to stay in the blood compartment. Therefore drugs which bind strongly to plasma protein tend to have lower distribution. • Of these plasma proteins, albumin, which comprises 50 % of the total proteins binds the widest range of drugs. Acidic drugs commonly bind to albumin, while basic drugs often bind to alpha1-acid glycoproteins and lipoproteins.
Apparent Volume of Distribution (Vd) • Definition:The apparent volume of distribution indicates into how large a volume the drug distributes if it were at the same concentration as that in plasma (or in other reference fluid which is sampled - blood, serum).
Apparent Volume of Distribution (Vd) • This apparent volume of distribution is not a physiological volume. It won't be lower than blood or plasma volume but it can be much larger than body volume for some drugs. It is a mathematical factor relating the amount of drug in the body and the concentration of drug in the measured compartment, usually plasma: Vd = AMOUNT of drug in the body CONCENTRATION in plasma
Apparent volume of distribution Vd = AMOUNT OF DRUG IN THE BODY CONCENTRATION IN PLASMA Vd = F x Dose - AMOUNT eliminated CONCENTRATION IN PLASMA F…. ..absolute bioavailability (bioavailable fraction of the dose) Units: volume (L, L/kg of TBW)
Rapid (bolus) i.v. injection and uniform mixing of the amount administered throughout the volume of total body water: Vd=Dose/cplasma Vd=Vtotal body water Vd= 0.6 L/kg BW Dose = cplasma . Vd Fat !!!0,2-0,35 L water per 1 kg of weight
Vd = Amount / Concentration in plasma • Most drugs are distributed unevenly into the body. Some drugs (digoxin) are extensively distributed and bound in tissues, leaving low concentrations in the plasma, thus the body as a whole appears to have a large volume of distribution.
Volumes of some compartments of the adult human body in relation to VD: Total body water 0.6 L/kg BW Intracellular water 0.4 L/kg BW Extracellular water 0.2 L/kg BW Plasma 0.04 L/kg BW VD 0.05 L/kg the drug remains in the blood (heparine) VD 0.1-0.3 L/kg distribution from blood into extracellular fluid (gentamicin - polar drugs). VD 0.6 L/kg distribution from blood into intracelular and extracellular fluid (methotrexate) VD>>0.6 L/kg distribution intracellularly and high binding in tissues (amiodarone - 350 L/kg)
Use of Vd: 1/Vd in conjunction with a target concentration CT can be used to compute a loading dose DL: DL = VD . CT
Loading dose = CP x VD L. Dose
Use of Vd: EXAMPLE: J.K.(TBW = 90 kg)was admitted to the ICU for pneumonia caused by Gram-negative bacteria. Calculate the loading dose of tobramycin for this patient to achieve the target average concentration of 4 mg/l. Tobramycin VD is 0.2 l/kg of TBW. Loading Dose = ? Loading Dose = 0.2 . TBW . Concentration Loading Dose = 0.2 . 90 . 4 = 72 mg
Use of Vd: 2) It can be usefull in case of overdoses and in certain medico-legal cases to estimate the amount of drug in the body: Amount in the body = Vd . Cactual, measured
Use of Vd: 3/ To assess feasibility of using hemoperfusion or dialysis for drug removal from the body: The larger the VD the smaller fraction of the dose is in plasma, the less is plasma concentration and the less efficient is any drug removal through extracorporeal mechanisms.
Clearance (CL) Definition : Clearance of a drug is the ratio of the rate of elimination by all routs to the concentration of drug in plasma. CL = Rate of eliminination[mg / h ] C in plasma[mg /L ] Unit: Volume/Time [L/h] or adjusted for body weight [l/h/kg]
Clearance (CL) Rate of eliminination = CL x C in plasma (Amount / Unit of time)= (Volume / Unit of time) x Cin plasma Unit: Volume/Time [L/h] Another possible way of understanding clearance: Clearance is the volume of plasma completly cleared of the drug per unit of time by all routes - by the liver, the kidney…).
Rate of elimination Elimination of most drugs from the body after therapeutically relevant doses follows first-order kinetics. To illustrate first order kinetics we might consider what would happen if we were to give a drug by i.v. bolus injection, collect blood samples at various times and measure the plasma concentrations of the drug. We might see a decrease in concentration as the drug is eliminated.
Rate of elimination Elimination which follows first-order kinetics: dC/dt = - kel . C kel ….rate constant of elimination rate of change is proportional to concentration and is therefore decreasing with time as the conc. decreases
Rate of elimination monoexponential decay: C(t) = C0 . e- kel . t half-life : t1/2 C= C0 / 2 t= t1/2 = ln2 / kel = 0.693/ kel after 4 half-lives: 6% remaining, 94% eliminated
Rate of elimination Elimination which follows first-order kinetics: semi-log graph. t 1/2 = 0.693/ kel kel can be estimated by means of the linear-regression analysis
Clearance (CL) Clearance has an additive character: it is the sum of clearences in all eliminating organs CL = CLRENAL + CLHEPATIC +CLpulmonary ...other renal + nonrenal
The principle of linear pharmacokinetics Linear (first-order) pharmacokinetics: For most drugs, clearance is constant over the plasma concentration range used in clinical practice. Elimination is not saturable (non-capacity-limited) and the rate of drug elimination is directly proporcionate to the concentration: Rate of elimin. = CL . Concentration
Nonlinear pharmacokinetics Nonlinear pharmacokinetics: (capacity-limited, dose or concentration dependent, saturable) CL varies depending on the concentration of a drug. Rate of elimination = Vmax . C /Michaelis- Menten/ Km + C CL = Vmax Km + C ethanol, phenytoin, theofylline
Use of clearance: 1/ Total clearance determines the average steady-state concentration of a drug during continuous drug administration (multiple intermittent dosing or constant rate i.v. infusion: at the steady-state: Rate of dosing = Rate of elimination = CL . Cpss continuous i.v. infusion: Cpss = Rate of inf./ CL
Use of clearance: 2/ Total clearance, when multiplied by a target steady-state concentration CSS,TARGET , can be used to calculate the dosing rate required to maintain plasma CSS,TARGET (the maintenance dose) Rate of dosing = CSS,TARGET . CL Rate of dosing ….. Rate of i.v. infusion (mg/h) ……Oral dose / Dosing interval …… i.v. dose / Dosing interval
Calculation of the maintenance dose J.K.was admitted to the ICU for pneumonia caused by Gram-negative bacteria. Calculate the maintenance dose (i.v.-infusion in 6 h intervals)of tobramycin for this patient to achive the target average concentration of 4 mg/l. Clearance of tobramycin was estimated to be 70 ml/min. Rate of dosing = Dose / Interval Rate of dosing = Rate of elimination = CL.cT Rate of dosing = 4.70.60 / 1000 = 16.8 mg/h Dose = Rate of dosing . Interval = 6 . 16.8 = 101 mg
Multiple i.v. bolus dose administration : drug accu- mulation in plasma until the steady state is achieved
Use of clearance: 3/ The numerical value of total clearance and its two principal components (hepatic and renal) provide important insights into the elimination processes and into the potential needs for dosage adjustments in case of liver or kidney impairment.
Biliary clearance CLh Q… hepatic blood flow per 1 min: 1.5 L/min, Q . Cin LIVER Q . Cout bile Amount excreted in bile = Amount extracted= Q.(Cin-Cout) CLh = rate of elimin. / Cin = Q . (Cin-Cout) / Cin CL= Q . E hepatic extraction ratio
Hepatic extraction ratio (E) E = (Cin-Cout) / Cin A/ High extraction: Cout 0, E 1 (>0.7) After oral administration, drug is efficiently extracted by the liver and less is available in the systemic circulation - high first-pass effect. The elimination of the drug from the systemic circulation is flow limited: (clearance = Q.E = Q). B/ Low extraction: Cout Cin , E 0 (<0.3) Small first-pass, high systemic availability after oral administration, hepatic clearance is sensitive to change in E (inhibition and induction of metabolism).
Renal clearance CLR GFR , C in plasma GFR100 -150 ml/min KIDNEY URINE VU , CU VU = volume collected / urine collection period Amount excreted = VU . CU CLR = rate of elim. / C in plasma= VU . CU/ C in plasma
Renal clearance CLR CLR = rate of elim. / C in plasma= VU . CU/ C in plasma Renal clearance of a drug is the ratio of the rate of elimination of the drug by the kidney divided by its concentration in plasma.
Renal clearance of the drug CLR = GFR x funbound + Tubular secretion - Tubular reabsorption Glomerular filtration rate is measured using endogenous creatinine: GFR creatinine clearance = 100 - 150 ml/min 1/ CLR > GFR x funbound: filtration + Tubular secretion 2/ CLR < GFR x funbound: filtration - Tubular reabsorption 3/ CLR GFR x funbound: filtration
Elimination half-life (t1/2) Definition:Elimination half-life is the time it takes the drug concentration in the blood to decline to one half of its initial value. It is a secondary parameter : The elimination half-life is dependent on the ratio of VD and CL. Unit : time (min, h, day)
Use of t1/2: 1/ t1/2 can be used to predict how long it will take for the drug to be eliminated from plasma.
Use of t1/2: 2/ t1/2 can be used to predict how long it will take from the start of dosing to reach steady-state levels during multiple dosing or continuous i.v. infusion. No. of t1/2 Concentration achieved (% of steady conc.) 1 50 2 75 3 87.5 4 94 5 97
Important During continuous (infusion) or continuous intermittent dosing (oral dosing): The steady-concentration depends on the rate of dosing (the dose/dosing interval) and the clearance. Time required to achieve steady-state depends on the half-life and is independent of the rate of dosing and the clearance..
Use of t1/2: 3/ the relationship between t1/2 and dosing interval t can be used to predict the degree of accumulation of a drug in the blood. The longer t1/2 and the shorter t, the more drug accumulates. t t1/2 Moderate accumulation during dosing 2-times) t < t1/2Significant accumulation during dosing (> 2-times) t > t1/2Insignificant accumulation during dosing (< 2-times)
Use of t1/2: 4/ t1/2 (the relationship between t1/2 and dosing interval t) can be used to predict the degree of fluctuation of a drug concentration within a dosing interval. t t1/2 Css,min levels at steady state are aprox. 50% of Css,max. Moderate fluctuation. t < t1/2Css,min levels at steady state are more than 50% of Css,max. Small fluctuation. t > t1/2 Css,min levels at steady state are less than 50% of Css,max. Wide fluctuation.
Multiple short i.v. infusions of amikacin: the rate of dosing is constant but interdose interval is changing, t1/2= 6 h
Multiple short i.v. infusions of amikacin: the rate of dosing is constant but interdose interval is changing, t1/2= 6 h