Download

1 / 12
160 likes | 1.12k Views
Ornithine Transcarbamylase Deficiency. Department of Neurosciences Canberra Hospital May 1999. Ornithine Transcarbamylase Deficiency.

E N D
Ornithine Transcarbamylase Deficiency Department of Neurosciences Canberra Hospital May 1999
Ornithine Transcarbamylase Deficiency • Ornithine transcarbamylase deficiency is an X-linked metabolic disorder involving the urea cycle. Consequences include hyperammonemia, hyperglutaminemia, hypoarginemia, hypocitrullinemia & episodic encephalopathy , that, if uncontrolled results in brain injury & death.
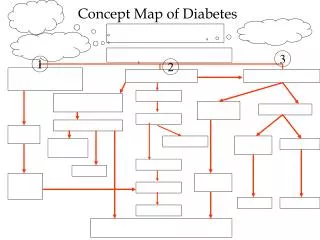
Pathophysiology (see urea cycle) • .Hyperammonemia: • Cerebral edema with subsequent raised intracranial pressure is primary • response to hyperammonemia. • Neurologic symptoms occur in absence of neuronal pathology. • .Hyperglutaminemia: • Strong relationship between hyperammonemia, neurologic dysfunction & • CSF glutamine concentration in hepatic encephalopathy.
Classical (Early onset) Presentation • Baby boy product of normal full term pregnancy with normal labor & no perinatal risk factors is normal for the first 24 hrs. • Over the ensuing days, worsening lethargy, requires stimulation for feeding; then develops problems with vomiting, hypothermia & hyperventilation. • A search for sepsis is unrevealing. • CT shows cerebral edema. • Death is inevitable unless early diagnosis & treatment commenced early. • Maybe a history of consanguinity, neonatal sib deaths, or neonatal male deaths on pedigree analysis.
Nonclassical Presentations • Genotypic & phenotypic variability • Lyonisation in females. • Environmental precipitants: • Sepsis • Postpartum • Protein loading • Post surgery • Drug induced in particular valproate.
Late Presentation • Both male & females. • Clinical features: • Vomiting • Lethargy, somnolescence, confusion. • Ataxia. • Amblyopia. • Aversion to protein (protein provoked headache). • Seizures.
Diagnosis • Normal LFT. • Initially normal CT. • EEG which may show diffuse encephalopathy. • UCE: low urea. • Mild respiratory alkalosis. • NH3 • Orotic acid in urine. • (arginine, citrulline, glutamine) • (liver biopsy with measurement of OTC activity)
Acute Treatment • Early diagnosis & management (delays are fatal) • Discontinue all iv & dietary nitrogen intake. • High parenteral caloric intake to prevent catabolism. • Monitor UCE, pH, Pco2, NH3. • Reduce raised intracranial pressure with osmotherapy (mannitol) not steroids. • Infusions of sodium benzoate, sodium phenylacetate & arginine. • Hemodialysis as emergency procedure if NH3 does not significantly decrease.
Long Term Treatment • To provide a diet sufficient in protein & energy to promote growth & development while avoiding metabolic disturbances ie protein restriction. • Sodium phenylbutyrate. • Avoidance of high risk situations or initiation of acute treatment of known OTC deficient patients who deteriorate in these situations (sepsis, peripartum, initiation of drugs) • Dialysis as possible long term measure.
Valproate & OTC Deficiency • 5 case reports in literature • First report in Lancet 1986. • 3/5 died even with correction of hyperammonemia. • Probably underrecognised. • Delay in correction of hyperammonemia in known OTC deficiency can also significantly increase mortality. • Many hypotheses concerning Valproate & OTC Deficiency (see chart). • Valproate can cause hyperammonemia in patients with no urea cycle defect.
Screening • Who should be screened: • Patients about to commence valproate with unexplained episodes of confusion especially in setting of known stress factors ie sepsis, postpartum. • Known family history of OTC deficiency. • Patients who have become confused on commencing valproate. • Family history of unexplained death in childhood (especially males) • Aversion to protein. • How should we screen? • UCE, LFT, NH3 level. • Provocative test such as allopurinol loading; not protein loading.
Summary • Beware of valproate induced hyperammonemic encephalopathy. • Measure UCE, LFT, NH3 urgently. • Cease valproate immediately. • Institute measures to reduce NH3 as delay increases mortality. • Report cases of valproate induced hyperammonemic encephalopathy. • Screen for urea cycle defect of which OTC deficiency is the commonest.