Download

1 / 52
520 likes | 653 Views
Strategies for Success with the IRB Tips to Help Get Your Protocol Approved. Susan S. Fish, PharmD, MPH Associate Director, Office of Clinical Research Director, Human Subjects Protection March 16, 2005. Tips to Help Get Your Protocol Approved. 1. Give the IRB what it needs

E N D
Strategies for Success with the IRBTips to Help Get Your Protocol Approved Susan S. Fish, PharmD, MPH Associate Director, Office of Clinical Research Director, Human Subjects Protection March 16, 2005
Tips to Help Get Your Protocol Approved 1. Give the IRB what it needs 2. Expedited vs Exempt 3. Study Procedure 4. Risks are Reasonable in Relation to Benefit 5. Sample Size 6. DSMP vs DSMB 7. Study Specific Justifications 8. Informed Consent 9. Attachments 10. Human Subjects Protection Certification 11. Amendments 12. Modification Memo Reply
1. Give the IRB What it Needs • Review Criteria • 45 CFR 46.111 • 21 CFR 56.111 • How do I know what the IRB needs?
Criteria For Approval 45 CFR 46.111 21 CFR 56.111 • Minimized risks • Reasonable risk/benefit relationship • Equitable subject selection • Informed consent process • Informed consent documentation • Data monitored for safety • Confidentiality/privacy maintained • Vulnerable populations protected
Take IRB Application Seriously • Allow enough time • Review spelling and grammar • Review for consistency and completeness • IRB only knows what the investigator is saying through what is in the application • Give the same attention and care to the IRB application as an NIH grant application
2. Expedited vs Exempt - Section B(English doesn’t work here!) Does this study involve greater than minimal risk to the subject? • If Yes (skip to Section C) • If No, then complete EITHER Section B1 or B2
Section B1- Exempt • For further information see Federal guidance for exempt categories (linked). • Common issues: • Anonymous (different from confidential) • Existing data • If Exempt, no need to complete Section J4 Waiver of Informed Consent • You do need HIPAA form(s)
SubjectMRNID Mary 1234567 001 Roz 2345678 002 Karen 3456789 003 IDRESULTS 001 534 002 723 003 421 Master Code List
SubjectMRN Mary 1234567 Roz 2345678 Karen 3456789 IDRESULTS 001 534 002 723 003 421 Anonymous Data
Section B2- Expedited • If charts/records not existing at time of IRB application • If master code list linking study ID with subject identifiers • If identity could be ascertained in “rich” database • If Expedited, must have either • Consent form – Section Q, or • Waiver of Informed Consent – Section J4
Confidentiality and Code List Samples will be collected and stored in the PI' s laboratory at the XXX, then will be shipped in batches to Laboratory of Dr. XXX for further analysis. Samples will be coded, and the code will be held by the PI. The list of the codes will be kept confidential and secured in password protected computer in locked office. The link to codes and names will be destroyed after the study completion and after the acceptance for publication, if appropriate.
3. Study ProcedureSection F2 • Describe in detail the experimental design, including all materials and all procedures to be performed in sequential order as they will be performed. Clarify which procedures/test articles are investigational and which are part of standard clinical care.
Research vs Standard of Care • Clearly describe standard of care (what would happen if subject does not consent to the research) • Clearly describe what is research • Caution re: therapeutic misconception • Research, not therapy or treatment • Researcher or Investigator, not doctor
Section F2:This description may include: • Methods • Specific information re: experimental interventions, such as dose and frequency of drug/placebo • Number, frequency and duration of subject contacts • Any additional requirements of the subject (post treatment follow-up, diary cards, questionnaires, etc.) • Outcomes (primary, secondary and how they will be measured) • Will any of the research be done outside BUMC (see A6a). • Entire duration of participation for a single subject • Estimated duration of recruitment and enrollment • Estimated duration of entire study (from initial IRB approval through data analysis to close of study) • Attach copies in Section S of any surveys, questionnaires, and other data collection instruments. Describe what is going to happen in the research. Tell us what you are going to do.
Study SummarySection C • 300 word summary in lay language • Describe study • Overview of study • Abstract • Used by reviewers
4. Risks are Reasonable in Relation to Benefit • Explain HOW the risks are outweighed by (or are reasonable in relation to) the benefits • Background- justify the statement of benefits • Consistency of risks section (H) with statement of minimal risk (B)
5. Sample SizeSection G • Please indicate why you chose the sample size proposed. • “Pilot” or Exploratory/Descriptive • Justify design regarding what conclusions will be drawn (i.e. feasibility, adverse effects) • Inform subjects of purpose • Sample size may be justified by the need to assess variability in the sample or to look for large effects
6. DSMP vs DSMBSection H • All studies > minimal risk need PLAN • Studies with substantial risk need BOARD • Risk • Subjective outcome, unblinded researchers • Some studies may need independent safety assessor, but not BOARD
Data Safety Monitoring Plan Indicate what measures will be taken to prevent or to minimize the effects of hazards, discomforts or inconveniences. Include a detailed description of your Data Safety Monitoring Plan (DSMP). The DSMP must minimally include • a description of the risks and safety assessments of the study, • specifics regarding who will be monitoring the data and the frequency of monitoring, and • a clear description of the safety findings that would cause the study to be suspended.
GCRC DSMP www.bumc.bu.edu/gcrc
Data Safety Monitoring Board • An independent group of experts who review accumulating study data for early indication of • Safety issues • Indication of efficacy • Futility • DSMB charter, SOPs, interim analysis rules, stopping rules
7. Study Specific Justifications • J3- Waiver of Documentation • J4- Waiver of Informed Consent, or Waiver of individual elements • O – FDA approved drug used off-label without IND
Waiver of DocumentationSection J3 If yes, please explain how your study meets one of the following two criteria [see 45 CFR 46.117(c)] : (1) That the only record linking the subject to the research would be the consent document and the principal risk of having a signed consent form would be potential harm resulting from a breach of confidentiality… [must have consent form in Section Q to offer subject]; or (2) That the research presents no more than minimal risk of harm to subjects and involves no procedures for which written consent is normally required outside of the research context.
Waiver of Consent or an ElementSection J4 • Explain how waiving the requirements for informed consent presents no more than minimal risk to study subjects. • Explain how waiving the requirements for informed consent will not adversely affect the rights and welfare of study subjects. • Explain why the research could not practicably be carried out without a waiver of informed consent or alteration of the informed consent process. • Provide a plan to disseminate pertinent information to subjects after the study is completed if consent is waived.
New Use of FDA Approved DrugSection O • If yes, insert the IND number in the IND box above and insert the drug/biological agent name, the name of the manufacturer, and who is holding the IND in the text box below. • If you feel that an IND is not required, justifyhow this study meets all of the criteria for 21CFR 312.2.A copy of the investigator's brochure or the drug package insert must be attached to Section S.
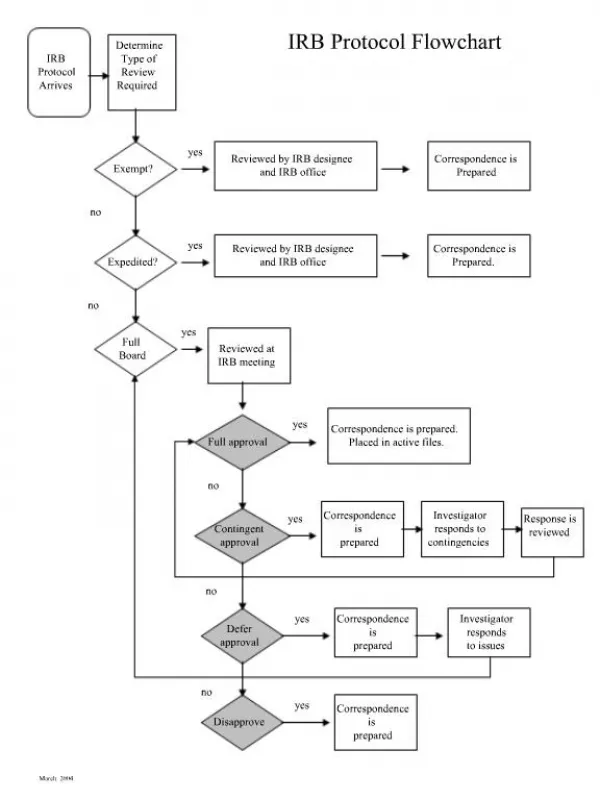
8. Informed Consent • Reading level too high, language legalistic • Needs extensive revision (put yourself in subject’s shoes) • Explain all procedures and risks • Explain implications rather than simply stating risks (i.e. liver function test abnormalities) • Flow chart as addendum?
Consent Form • Copy into Word for • Spell check • Grammar check • Readability
How to Check Readability-1 • Tools • Spelling and grammar • Options • Show readability statistics • Flesch-Kincaid grade level • Syllables per word • Words per sentence
9. Attachments – Section S • Use Appropriate Category • Only attach: .doc .jpg .pdf .xls
Attachment Categories-1 • Back translation #1-4 • Consent form previously approve #1-6 • Device brochure, and version 2 • Drug brochure, and version 2 • Drug enclosure, and version 2 • Educational materials #1, 2 • Final letter #1 • Follow up letter #1
Attachment Categories-2 • Foreign language consent form #1-4 • FWA/IRB approval letter from another institution #1-3 • Grant application • Informational brochures • Instruction sheet(s) #1,2 • Letter(s) to subjects #1, 2 • Medical record release form(s) • Note to IRB office #1-4
Attachment Categories-3 • Other #1-20 • Recruitment materials #1,2 • SAE at BUMC #1, 2 • Safety reports #1 • Section F- Investigator’s detailed protocol • Section F2 – GCRC DSMP • Section F2 –DSMP • Section F2 -- Procedure
Attachment Categories-4 • Section F3: Sponsor’s protocol • Section K: Certificate of Confidentiality • Section K2: HIPAA form(s), and #2 • Spanish Consent #1-3 • Subject contact letter #1-4 • Survey instrument/questionnaires #1, 2 • VA consent form #1-4
10. Are You Certified in Human Subjects Protection? www.bumc.bu.edu/irb www,bumc.bu.ed
11. Amendments • Summarize major changes in amendment • State if change in risk to subjects • State if change in consent form • Attach details of amendment • Revise IRB protocol to reflect all aspects of amendment. • Be sure that all changes are stated either in cover sheet or attachment.
12. Modification Memo Reply • IRB’s statement PI’S RESPONSE 2. IRB’s statement PI’S RESPONSE 3. IRB’s statement PI’S RESPONSE • Make changes in IRB protocol , ICF
Do’s and Don’t’sRichard Saitz, MD, MPH 2001 • If not written down the Board doesn’t know it (IRB cannot make [best or worst case] assumptions) • All protocol issues need to be in the protocol (i.e. not just in the ICF) • Address regulations explicitly • Tell the IRB what is part of the research and what is not, explicitly • Don’t try to bypass the process (i.e. don’t send directly to Chair); materials will get lost, delayed • Specifically define inclusion and exclusion criteria
Do’s and Don’t’sRichard Saitz, MD, MPH 2001 • Be clear when a study is not testing a treatment, not testing treatment efficacy • Avoid cut and paste errors (very common) and checking off the wrong boxes • Be explicit when study will pay for care, and particularly regarding payment for screening procedures for ineligibles • Be explicit about differences between sponsor protocol/grant proposal and local study procedures • If research lab used, address any delays
Do’s and Don’t’sRichard Saitz, MD, MPH 2001 • Avoid typographical errors • Be explicit about why a drug study is exempt from an IND • Consistency throughout application • Avoid saying what has been approved in the past • Avoid asking for speedy review (whose protocol should be delayed)?