Download

1 / 44
440 likes | 715 Views
Protein Purification February 5 2003. “A basic comprehension of the methods described here is necessary for an appreciation of the significance and the limitations of the information presented in the text ”. Protein Isolation. Must have sensitive method for detection.

E N D
“A basic comprehension of the methods described here is necessary for an appreciation of the significance and the limitations of the information presented in the text”
Protein Isolation • Must have sensitive method for detection. • Select a good source for the protein. • a. Rich source of material. • i.e. Heart mitochondria for cytochrome C • b. baker’s yeast (Saccharomyces cerevisiae) • c. Escherichia coli (recombinant expression) • Tissue specificity: Brain vs. kidney vs. eye. • Chickens, cows, pigs or rats are often used. • Molecular cloning techniques have allowed biochemists to over-express desired proteins in bacteria or C.H.O. (Chinese Hamster Ovary) cells by isolating the gene and placing it into a host system.
Methods of solubilization animal cells • Cells can be lysed by hypotonic shock. • Cells with high salt inside and no salt outside will • swell and rupture • Bacteria outer membranes must be digested. • Gram-negative bacteria • Hen egg white lysozyme digests b (1-4) linkages in the (glycosidic bonds) of polysaccharides. • Mechanical breakage blenders homogenizers • French press - high pressure 20,000 lbs/in2 forced through a small hole disrupts cells • ultrasound or sonication disrupts cells.
Centrifugation • Lysate - broken (lysed) cells- can be separated using • differential centrifugation • RPM - “spun down” • separates by density differences or by size (MW) of particles. • Cellular fractionation can separate: • mitochondria • microsomes • ribosomes • soluble proteins
Centrifugation: Units Where: w = angular velocity v = velocity of particle R = distance from center of rotation M = molecular weight V = partial specific volume of particle r = density of solvent Sedimentation velocity (Svedberg Coefficient) S = s x 10-13
Stability: proteins can denature!! H-bonds, ionic bonds, Van der Waals interactions, and Hydrophobic interactions can be disrupted. Denaturation is the process by which a protein loses its “native” or active shape or conformation. Temperature can play a role “cold labile” “heat labile” Protect against-Proteases, Inhibitors, Changes in pH, Protein can be air-denatured -egg white meringue - absorption to surfaces Damaged by oxidation 02 Heavy and transition metals damage proteins -they bind to protein- Cu+ Hg+ Bacterial contamination can destroy the protein
Activity Measurements In order to follow the purity of an enzyme, you need a method to measure its activity. Spectraphotometric analysis- is one common method to measure activity. Substrate [S] Product [P] a change of [S] with time if S is colored “absorbs light” we can use Beer’s Law. A = eb c c - concentration e -millimolar extinction coefficient A - absorbance b - path length T - percent transmittance A = - log % T if A then c at max
enzyme For the reaction: NADH NAD+ + H- NADH l Max = 340 nm DA Absorbance NAD+ 300 nm 350 nm Volume is 1 ml so micromoles NADH oxidized } = Specific activity DT min mg mg of protein
Start with one liter of lysed cells. We measure the rate of .01 ml of cells at at concentration of 20 mg/ml. i.e. the amount of enzyme we will assay is 0.01 ml We get a rate of A = 0.5 A/min 1 millimolar = 6.22 DA = e mM 0.5/6.22 = .008 millmolar/min and our assay volume = 1 ml 1 millimolar in a volume of one ml = 1 micromole/ml = mole C=.008 moles in 1 ml/min = .04 moles 0.2 mg min/mg
Total activity: .04 mmoles x 20 mg/ml = 0.8 mmoles / ml 0.8moles x 1000 ml = 800moles in 1 liter of cells ml min Red = is our enzyme If we remove greens & blues the specific activity increases, however, our total activity remains the same. If We lose red the total activity decreases.
We usually monitor both the total activity and specific activity for each purification step. Until the Specific Activity reaches a maximal value. How do we know if it is pure? Usually SDS - Page See Table 5-4 in Voet and Voet Some enzymes have no easy assay but the product of the reaction can be used in another reaction: enz1enz2 A B C NADH NAD+ Coupled Reactions: We couple enz2 to enz1 and measure NADH to get A
Use of radioactivity ATP ADP + Pi Separate ATP + Pi + ADP on TLC measure radioactivity Phosphoimager makes this easy else cut spots and count in scintillation counter. Pi ATP
Strategy of Purification • Fractionation procedures or steps to isolate protein based on physical characteristics. • Characteristic Procedure • Charge 1. Ion exchange • 2. Electrophoresis • 3. Isoelectric focusing • Polarity 1. Adsorption chromatography • 2. Paper chromatography • 3. Reverse phase chromatography • 4. Hydrophobic interaction
Characteristic Procedure • Size 1. Dialysis and ultrafiltration • 2. Gel electrophoresis • 3. Gel filtration • 4. Ultracentrifugation • Specificity 1. Affinity chromatography • 2. Immunopurification • Solubility 1. Salt precipitation • 2. Detergent solubilization
Ionic Strength • Ci = the molar concentration of the ith species • Zi = it’s ionic charge • 1M Na+ Cl- Z = 1 Na+ Z = 1 Cl- • 1 = (1M x 1)Na + (1M x 1)Cl • 2
For di- or tri-valent ions, where I is different than M • 1M MgCl2 • Mg++ = 1M, and Z = 2 • while Cl- = 2M, and Z =1 • I = (1 x 22)Mg + (2 x 12)Cl = 4 + 2 = 3 • 2 2
Salting out Use (NH4)2 SO4 : it is a Very Soluble salt that does not harm proteins. Refer to the Hofmiester Series
Chromatography • Analytical methods used to separate molecules. Involves a mobile and a stationary phase. • Mobile phase is what the material to be separated is dissolved in. • Stationary phase is a porous solid matrix which the mobile phase surrounds. • Separation occurs because of the differing chemistries each molecule has with both the mobile and stationary phase. • Chemistries are different depending on the specific method.
Types of chromatography • Gas - Solid: Mobile phase is gaseous, stationary phase is a solid matrix. • Liquid - Solid: Mobile phase is liquid, stationary phase is a solid matrix. • If separation is based on ionic interaction the method is called Ion Exchange chromatography. • If separation is based on solubility differences between the phases the method is called adsorption chromatography. • If the separation is base on size of molecule the method is called gel filtration or size exclusion. • If the separation is base on ligand affinity the method is called Affinity chromatography.
Ion ExchangeChromatography • A solid matrix with a positive charge i.e. R+ can bind different anions with different affinities. • We can swap one counter ion for another • (R+A-) + B- (R+B-) + A- • R = Resin and exchanges Anions (-) • This is an anion exchange resin. • There are also cation exchange resins. The type of an R group can determine the strength of interaction between the matrix, R and the counter ion. • If R is R- • (R-A+) + B+ (R-B+) + A-
Proteins have a net charge. The charge is positive below pI, while the charge is negative above pI The choice of exchange resin depends on the charge of the protein and the pH at which you want to do the purification. Once the protein binds, all unbound proteins are washed off the column. Bound proteins are eluted by increasing the ionic strength, changing the counter ion or changing the pH altering the charge on the protein or the column.
Paper chromatography • Stationary phase vs.. the Mobile phase • Partitioning between the two phases • Partition coefficient • The more H2O soluble the slower it migrates. • The more organic soluble the more it migrates. • The aqueous component of the solvent combines with the cellulose of the paper and becomes the stationary phase.
Materials can be visualized by: • Radioactivity • Fluorescence • UV absorbency • Stained with one of several dyes • Ninhydrin • Iodine • Sulfuric acid
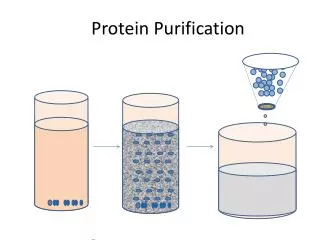
Gel FiltrationSize exclusion • A matrix with holes in it. • Vt = Vx + Vo • Vo = void volume = volume outside the “caves or knooks and crannies” • Vx occupied by gel beads • Vo 35% of Vt
Gel filtration can be used to determine the molecular mass of proteins • Ve = elution volume Vo = exclusion volume • Common matrix: dextran, agarose, or polyacrylamide • also desalts proteins
Before swelling the dry bead size 5% of Vt 60% are “holes” Hole sizes can be made different Small molecules see a larger column volume than big molecules and they get hung up in the caves. Large proteins are excluded, while small protein are included. Separation on size and shape.
Dialysis is a process that separates molecules according to size through the use of semipermeable membranes containing pores of less than macromolecular dimensions
Affinity Chromatography Based on molecular complementary between an enzyme and substrate. The substrate (R) is linked to a matrix with a spacer arm Only protein that binds R will stick to column. put citrate on column citrate dehydrogenase will specifically bind. Add excess citrate and the enzyme will be released.
The purification of Staphylococcal nuclease using the ligand, diphosphothymadine
Electrophoresis The migration of ions in an electric field Fele = qE where q is the charge and E is the electric Field strength Opposing this is Ffriction = vf where v = velocity of migration f is the frictional force. qE = vf
Separates on charge and size pH matters as well as the pI of the protein. Can be run at several pH values depending on proteins. DNA can also be separated on agarose gels. Genomic sized DNA can also be separated but requires more sophisticated equipment.
Proteins can be visualized by several methods Stained with a Dye: Coomassie blue Fluorescamine stain for fluorescence Silver staining very sensitive proteins can be labeled with radioactivity and visualized by exposure to X- ray film
SDS-PAGE Add sodium dodecyl sulfate, a 12 carbon detergent to give a negative charge to the protein. SDS also denatures the protein and collapses into a globular ball. The proteins are separated by molecular mass