INTERSTITIAL PULMONARY FIBROSIS
560 likes | 995 Views
INTERSTITIAL PULMONARY FIBROSIS. By: Dr.Bidhi Chand. Junior Resident Pulmonary Medicine. INTERSTITIAL PULMONARY FIBROSIS ATS Definition

INTERSTITIAL PULMONARY FIBROSIS
E N D
Presentation Transcript
INTERSTITIAL PULMONARY FIBROSIS By: Dr.BidhiChand Junior Resident Pulmonary Medicine
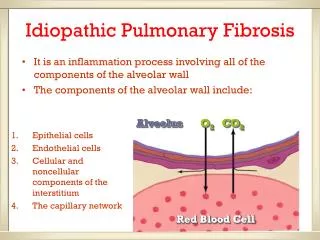
INTERSTITIAL PULMONARY FIBROSIS ATS Definition Interstitial Pulmonary Fibrosis is defined as a specific form of chronic fibrosing interstitial pneumonia of unknown causes, limited to the lungs and associated with a histologic pattern of usual interstitial pneumonia (UIP).
EPIDEMIOLOGY • Estimated to affect approximated 5 million people world wide. • The most common (and deadly) interstitial lung disease. • Most cases are sporadic, but rare cases of familial IPF have been described. • 15 % of pulmonary physician’s practice. • Mortality from IPF continues to increase in many countries. • Earlier, considered to be rare in India. • In 1979, Jindal et al published 65 cases of DPLD seen over a period of 5 years. Recently the same center published data on 76 patients with IPF diagnosed over a 16 months period- increase in frequency of diagnosis. • Increase in number of studies in India due to increase in incidence or due to availability of better diagnostic facilities like HRCT and fibreopticbronchoscopy.
RISK FACTOR OF IPF • Cigarette smoking. • Environmental Exposure – metal dust, wood dust, solvents associated with increased risk of developing pulmonary fibrosis. • Viruses (EBV, influenza, CMV, HCV), chronic aspiration secondary to GERD. • Hereditary factors do contribute but no specific genetic marker has been identified. • Familial IPF – autosomal dominant – variable penetrance.
PATHOLOGY • Interstitial pneumonia classified by Hammanand Rich followed by Liebow. • Pathological Classification • Usual interstitial pneumonia. • Desquamative interstitial pneumonia. • Respiratory BronchiolitisInterstitial Lung Diseases. • Acute Interstitial pneumonia (Hamman-Rich Disease) • Non-specific Interstitial pneumonia.
PATHOLOGY (Contd.) • Pathological changes characterized by UIP – distinguished by variation in location and age of lesions, with predilection for peripheral subpleuralparenchyma. • Fibrotic zones with associated honey-combing alternate with areas of relatively unaffected Lung tissue. Fibrotic areas vary in age and activity.
Multiple Hypotheses for the Pathogenesis of IPF • Inflammation causes fibrosis • Noninflammatory (multiple hit) hypothesis: fibrosis results from epithelial injury and abnormal wound healing in the absence of chronic inflammation • Vascular remodeling: aberrant vascular remodeling supports fibrosis, and may contribute to increased shunt and hypoxemia
Inflammatory Hypothesis • Inflammation causes fibrosis • Inflammatory concept was dominant in the 1970s and 1980s • IPF resulted from unremitting inflammatory response to injury culminating in progressive fibrosis • Role of inflammation remains controversial • Lack of efficacy of corticosteroids Injury Inflammation Fibrosis
Progression of Lung Fibrosis Injury Epithelial cells ? Endothelial cells Capillary
Tissue Model of Lung Fibrosis Cell death Epithelial cells Endothelial cells Growth factors and other products of epithelial cell Injury Capillary Myofibroblast Collagen
Non inflammatory (multiple hit) Hypothesis • Fibrosis results from epithelial/endothelial injury and abnormal wound healing in the absence of chronic inflammation • Recurrent, unknown injury to distal pulmonary parenchyma causes repeated epithelial cell injury and apoptosis • Loss of alveolar epithelium exposes basement membrane to oxidative injury and degradation • Failure of re-epithelialization/re-endothelialization provides stimulus for persistent profibrotic growth factor production, persistent fibroblast proliferation, excessive deposition of ECM, and progressive fibrosis
Noninflammatory (multiple hit) Hypothesis Recurrent pulmonary injury Epithelial/ endothelial injury and apoptosis TGF-b = transforming growth factor-beta PDGF = platelet derived growth factor IGF-1 = insulin-like growth factor-1 Loss of basement membrane Failure of re-epithelialization/ re-endothelialization Release of profibrotic growth factors (TGF-b, PDGF, IGF-1) Fibroblast proliferation ECM deposition Progressive fibrosis with loss of lung architecture
Vascular Remodeling Hypothesis • Aberrant vascular remodeling supports fibrosis and may contribute to increased shunt and hypoxemia • Increased angiogenesis results from imbalance of pro-angiogenicchemokines (IL-8, ENA-78) and anti-angiogenic, IFN-inducible chemokines (IP-10) • Vascular remodeling leads to anastomoses between the systemic/pulmonary microvasculature, increasing right-to-left shunt, contributing to hypoxemia Fibrosis Chemokine imbalance Increased angiogenesis Aberrant vascular remodeling
Defects in Host Defense Mechanisms May Contribute to Fibrosis • Defects in endogenous host defense mechanisms (eg, IFN-g, PGE2 production) that limit fibrosis after acute lung injury may contribute to progressive fibrosis
American Thoracic Society (ATS) • Diagnostic Criteria (IPF) • According to ATS, in immunocompetent adult, the presence of all of the major diagnostic criteria as well as at least 3-4 minor criteria increase the likelihood of correct diagnosis of IPF.
American Thoracic Society (ATS) • Major Criteria • Exclusion of other known causes of ILD such as certain drug toxicities, environmental exposures and connective tissue disorders. • Abnormal pulmonary function studies that include restriction (reduced VC, increase FEV1 / FVC ratio) and impaired gas exchange. • Bibasilar reticular abnormalities with minimal ground glass opacities on HRCT scan. • TransbronchialLung biopsy or BAL showing no feature to support alternative diagnosis.
Minor Criteria • Age > 50 years. • Insidious onset of otherwise unexplained dyspnoea on exertion. • Duration of illness more or equal to 3 months. • Bibasilar, inspiratory crackles (dry/velcro type in quality)
CLINICAL PRESENTATION • Middle age 50 - 70. • Progressive dyspnoea on exertion. • Non-productive cough. • Most have symptoms for 12-18 month prior to definitive evaluation. • Constitutional symptoms are uncommon. • Weight loss, fever, fatigue, myalgias, or arthralgiasoccasionally present.
PHYSICAL EXAMINATION • Bibasilar late inspiratory fine crackles (velcrorales). • Tachypnea. • Clubbing 40-70% late in disease course. • Cardiac examination normal until middle-late stages – augmental P2, Right sided heave, S3 gallop. • Cyanosis
Diagnosis of Pulmonary Fibrosis • Investigation: • FBC show mild anemia. • ESR and C-reactive protein may be raised. • Autoantibodiesincluding antinuclear antibodies, RA factors. • Arterial blood gases – Oxygen desaturation is common. • Lung Function Test : • Shows Restrictive Pattern (But obstructive of airway may also be present) • Reduced total lung capacity. • Reduced residual capacity. • Reduced residual volume. • Reduced gas transfer.
X-ray Chest • Bilateral basal symmetrical peripheral reticular opacities with decreased lung volumes. • Volume loss evident by diaphragmatic elevation and depression of fissures. • Progressive fibrosis ultimately leads to cystic dilatation of distal air spaces, which is visible as peripheral honey combing. • Alternative diagnosis/ super-imposed complicated illness should be suspected if X-ray shows bronchogram, confluent shadows or hilaradenopathy. • Most of patient with IPF have abnormalities on X-ray. Rarely patients can have a normal X-ray, but have evidence of IPF either in HRCT or surgical lung biopsy.
HRCT • Patchy, predominantly peripheral, sub-pleural, bibasilar reticular abnormalities and area of traction bronchiectasis with limited amount of ground glass opacity. • The areas of severe involvement show sub-pleural honeycombing. • When certain diagnosis is not able to made by HRCT then a lung biopsy is needed for diagnosis.
This 50-year-old man presented with end-stage lung fibrosis PA chest radiograph shows medium to coarse reticular B: CT scan shows multiple small cysts (honeycombing) involving predominantly the subpleural peripheral regions of lung. Traction bronchiectasis, another sign of end-stage lung fibrosis.
Classic idiopathic pulmonary fibrosis in 70-year-old man. High-resolution CT shows bilateral subpleural reticulation, traction bronchiectasis (curved arrow), and honeycombing (straight arrows).
42-year-old woman with biopsy-proven idiopathic pulmonary fibrosis. High-resolution CT shows patchy bilateral ground-glass opacities and mild predominantly subpleural reticulation.
57-year-old man with biopsy-proven idiopathic pulmonary fibrosis. High-resolution CT (HRCT) shows patchy bilateral areas of ground-glass opacity. Fine reticulation is observed in subpleural regions.
BRONCHOALVEOLAR LAVAGE • Increase in Polymorphonuclear leucocytes, neutrophil products, eosinophils, activated alveolar macrophage, alveolar macrophages products, cytokines, growth factors. • BAL-useful research tool but its diagnostic usefulness is limited. • Increase in neutrophils/ eosinophils in BAL fluid – worse prgonosis. • BAL lymphocytosis – less honey combing and greater responsiveness to treatment.
TBLB (Transbronchial Lung Biopsy) Not helpful in making a diagnosis of IPF as limited Lung tissue is obtained. But can exclude by identifying an alternative specific diagnosis.
Surgical Lung Biopsy By open or video assisted thoracoscopic methods – gold standard for diagnosis of IPF. Large piece of Lung parenchyma is required, optimally from several sites.
USUAL INTERSTITIAL PNEUMONIA PATTERN Mason: Murray & Nadel's Textbook of Respiratory Medicine, 4th ed. Idiopathic Pulmonary Fibrosis, Gross and Huninghake, NEJM, 2001.
COMPLICATIONS OF IPF Interstitial lung disease can lead to series of life-threatening complications, including :- Pulmonary Hypertension Unlike systemic high Blood Pressure, this condition affects only the artries in Lungs. It begins when scar tissue retricts the smaller blood vessels, limiting blood flow in Lungs. This in turn raises pressure within the Pulmonary arteries. Pulmonary hypertension is a serious illness that becomes progressively worse. Cont…..
Right sided heart failure (cor-pulmonale) : This is serious condition occur when heart’s lower chamber (right ventricle) : which is less muscular than the left- has to pump harder than usual to move blood through obstructed pulmonary arteries Eventually the right ventricle fails from the extra strain. • Respiratory failure : In the end stage of chronic interstitial lung disease, respiratory failure occurs when severely low blood oxygen levels along with rising pressure in the pulmonary arteries and right ventricle causes heart failure.
The Course of IPF Although the course of idiopathic pulmonary fibrosis varies greatly from person to person, the disease usually develops slowly, sometimes over years. The early stages are marked by alveolitis, an inflammation of the air sacs called alveoli, in the lungs. The job of the air sacs is to allow the transfer of O2 from Lungs into the blood and elimination of CO2 from Lungs and out of the body. Contd…
As IPF progresses, the alveoli become damaged and scarred, the stiffening of the lungs, makes breathing difficult and bring on a feeling of breathlessness, especially during activities that require extra effort. In addition, scarring of the alveoli reduces the ability of lungs to transfer oxygen. The resulting lack of O2 in to the blood (hypoxemia) may cause increase in the blood vessels of the lungs, a situation known as pulmonary hypertension. The high blood pressure in the lungs then puts a strain on right ventricle the lower right side of the heart, which pumps the oxygen – poor blood into the lungs.
TREATMENT AND DRUGS • The Lung Scarring that occurs in pulmonary fibrosis cannot be reversed, and no current treatment has proved effective in stopping the ultimate progression of the disease. Some treatments, may improve symptoms temporarily or slow the disease’s progress. Other improve quality of life. Cont…..
Specific Treatment Options are: • Medications • Oxygen Therapy • Pulmonary Rehabilitation • Lung Transplantation Cont…..
Medications : • Corticosteroids • Immunosupressant/ Cytotoxic agents • Antifibrotic agents in along or combination • Anti-oxidant agents Cont…..
Corticosteroids: • The main stay of treatment for idiopathic pulmonary fibrosis is medication that help to reduce inflammation. Inflammation is thought to play a role in the injury and fibrosis of Lung tissue. So corticosteroids that stop the inflammation are thought to have an effect on the resulting damage. • Despite their widespread use only 10-30% of pateint with IPF improve on quantitative assessment when treated with corticosteroids. Cont…..
If response is to occur, it is usually noted with three months. • Prolonged treatment for a minimum of 1-2 years and sometimes indefinitely is reasonable for patient exhibiting unequivocal response to therapy. • High dose IV pulse methylprednisolonehas no proven advantage over oral corticosteriods. • Now, treatment with steroid alone is considered in appropriate, corticosteroids, should be used in conjuctionwith cytotoxic agents. • Favorable response to combination is seen in 15-50% of cases. Cont…..
CYTOTOXIC / IMMUNOSUPPRESSANT Azothioprine Cyclophosphamide Azothioprine : is a medicine that effects the immune system. Because it can cause serious side effects, it may be prescribed with corticosteroids – is associated with improvement and enhanced survival in some patient. Cyclophosphamide : High dose IV administered every 2-4 weeks (500-1800mg) – tried in open trial result were un-impressive. A recent study suggested that combined corticosteroid and cyclophosphamide therapy has no impact on the survival of patient with IPF.
AGENTS THAT AFFECT COLLAGEN SYNTHESIS OR FIBROSIS Colchicine D-Pencillamine Interferon Gamma Pirfenidone Interferon gamma : inhibits proliferation of lung fibroblasts in dose dependent manner and a reduce protein synthesis in fibroblasts. - In an open randomized study with 18 patients who had not respondendto glucocorticoidsand other immunosupressive agents – 200mg interferon gamma 1b , 3 times/wk for 12 month along with predinsolone resulted in improvement in TLC and partial pressure of O2. Cont…..
Subsequent studies dampened hopes, when Honore et al reported 4 cases of IPF who developed irreversible respiratory failure following treatment with Interferon Gamma. • Increased level of IL-18 in induced sputum of patient with IPF have been found to decrease after treatment with interferon gamma. • A recent meta analysis showed interferon therapy – reduced mortality.
Pirfenidone – Anti Fibrotic Agents A larged well controlled multinational clinical trial programme has been demonstrated the effectiveness and safety of pirfenidone in the treatment of IPF. Study present in American Thoracic Society International Conference, Tronoto. Daily consumption of pirfenidonecan slow down the progression of IPF by improving the lung capacity. Study conducted on 275 patient by Takashi Ogura in Japanese patients with mild-moderate IPF patient was randomly divided into three groups were administered 1800mg/ day (High dose) or 1200mg/day (low dose) of pirfenidone or placebo for 52 weeks. Cont…..
Then vital capacity assessed at week 52, the researchers found that the loss of vital capacity in high dose regimen group was significantly lower when compared with the low dose regime and with the placebo group. The rate of deterioration IPF was reduced in patients taking pirfenidone. The drug was relatively well tolerated. The only side effect of the drug to be noticed was photosensitivity, which as mild severity.
Anti Oxidant Agents • Glutathione , taurine, niacin, inhibit development of fibrosis in animal models. • Acetylecysteine – a precurser of glutathione, can replenish pulmonary glutathione levels. • In study with 18 patients with IPF treated with 600mg N-acetyl cysteine TID for 12 weeks in addition to their latest immunosupressant therapy – significant improvement PFT. • A recent double blind, randomized, placebo controlled multicenter study assessed the effectiveness over 1 year of high dose oral acetyl cysteine added to standard therapy with prednisolone and azothioprine – show acetyl cysteineslowed deterioration of vital capacity at the end of 1 year. Cont…..
Pulmonary Rehabilitation / General Supportive measures : The aim of the pulmonary rehabilitation is not only to improve daily functioning, but also to help people with interstitial lung disease to live fully satisfying life. Pulmonary rehabilitation program focus on: Physical exercise, to improve your endurance. Breathing techniques that improve lung efficiency. Emotional Support Nutritional Counselling
General Supportive measures : • Smoking cessation • Avoidance of any other environmental (including occupational) causes. • Stopping any medication thought to be causing or contributing to pulmonary fibrosis. • Treating any respiratory tract infections promptly. • Patients should be encouraged to receive influenza or vaccination and pneumococal vaccine. • Good pulmonary hygiene is important.
Lung Transplantation : Lung transplantation is a treatment option for selected patients with advanced disease refractory to medical therapy. Lung transplant improves long term survival. Survival rates worldwide after single lung transplantation are approximately : 74% at 1 year 58% at 3 years 47% at 5 years and 42% at 10 years
PROGNOSIS : The prognosis variable and depends on the specific diagnosis and severity. Some disease are insidious in onset and gradual progression, while other disease are acute in onset but responsive to therapy. Idiopathic pulmonary fibrosis is progressive illness, producing increasingly severe symptoms, and generally has a poor prognosis. Mortality data for 3 year and 5 year mortality rate are approximately 50% and 80% respectively. Although IPF occurs in older patients with co-morbid diseases, most patients with IPF die as direct consequence of the Lung fibrosis. However, some patients with IPF remain stable for a number of years. The median survival rate of biopsy – proven IPF is less than three years. Most will die as a result of respiratory failure, but other will develop infections secondary to steroid therapy or right heart failure.