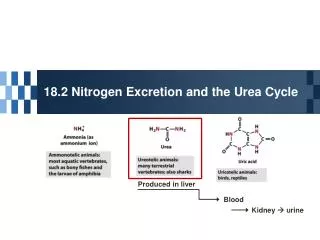
UREA CYCLE
UREA CYCLE. O. H. N. HN. O. N. N. O. H. H. most terrestrial vertebrates. fish & other aquatic vertebrates. birds & reptiles. O. H 2 N-C-NH 2 urea. amino acids. The carbon chains are broken down to molecules that feed into the TCA cycle.


UREA CYCLE
E N D
Presentation Transcript
O H N HN O N N O H H most terrestrial vertebrates fish & other aquatic vertebrates birds & reptiles O H2N-C-NH2urea amino acids The carbon chains are broken down to molecules that feed into the TCA cycle. Most mammals convert amino-acid nitrogen to urea for excretion NH4+ Some animals excrete NH4+ or uric acid. NH4+ ammonium ion uric acid
Ammonia is a toxic substance to plants and animals (especially for brain) Normal concentration:25-40 mol/l (0.4-0.7 mg/l) Ammonia must be removed from the organism Terrestrial vertebrates synthesize urea (excreted by the kidneys) - ureotelic organisms Urea formation takes place in the liver Birds, reptiles synthesize uric acid
Why Urea? • Non toxic • Water soluble • Combines two waste products into one molecule: • CO2 • NH3
Ammonia is highly toxic • Main reason to form urea is to reduce levels of ammonia • “Ammonia” often refers to (NH3 + NH4+) • NH3 is really ammonia • NH4+ is the ammonium ion
Hypotheses toxicity of ammoniaA. The binding of ammonia in the synthesis of glutamate causes an outflow of α-ketoglutarate from the tricarboxylic acid cycle, with decreased formation of ATP energy and deteriorates the activity of cells.B. Ammonium ions NH4 + caused alkalization of blood plasma. This increases the affinity of hemoglobin for oxygen (Bohr effect), the hemoglobin does not release oxygen to the capillaries, resulting the cells hypoxia occurs.C. The accumulation of free NH4 + ion in the cytosol affects the membrane potential and intracellular enzymes work - it competes with ion pumps, Na + and K +.
Hypotheses toxicity of ammoniaD. The producing ammonia tramsform glutamic acid - glutamine - an osmotically active substance. This leads to water retention in the cells and the swelling that causes swelling of tissues. In the case of nervous tissue it can cause brain swelling, coma and death.E. The use of α-ketoglutarate and glutamate to neutralize the ammonia causes a decrease in the synthesis of γ-aminobutyric acid (GABA) inhibitory neurotransmitter of the nervous system.
AMMONIA METABOLISM The ways of ammonia formation 1. Oxidative deamination of amino acids 2. Deamination of physiologically active amines and nitrogenous bases. 3. Absorption of ammonia from intestine (degradation of proteins by intestinal microorganisms results in the ammonia formation). 4. Hydrolytic deamination of AMP in the brain (enzyme – adenosine deaminase)
Glutamate is not deaminated in peripheral tissues Peripheral Tissues Transport Nitrogen to the Liver Two ways of nitrogen transport from peripheral tissues (muscle) to the liver: 1. Alanine cycle. Glutamate is formed by transamination reactions
Nitrogen is then transferred to pyruvate to form alanine, which is released into the blood. The liver takes up the alanine and converts it back into pyruvate by transamination. The glutamate formed in the liver is deaminated and ammonia is utilized in urea cycle.
Closer look at transport of waste N from peripheral tissue to liver via alanine and glutamine Waste N funnelled to pyruvate via transaminations Glucose – Alanine Cycle Net: N (muscle) Urea (liver)
2. Nitrogen can be transported as glutamine. Glutamine synthetasecatalyzes the synthesis of glutamine from glutamate and NH4+ in an ATP-dependent reaction: Ammonia transport in the form of glutamine. Excessammonia in tissues is added to glutamate to form glutamine, a processcatalyzed by glutamine synthetase. After transport in the bloodstream,the glutamine enters the liver and NH4 is liberated in mitochondriaby the enzyme glutaminase.
Synthesis of Glutamine in Peripheral Tissue and Transport to the Liver
Overview • Occurs primarily in liver; excreted by kidney • Principal method for removing ammonia • Hyperammonemia: • Defects in urea cycle enzymes (CPS, OTC, etc.) • Severe neurological defects in neonates • Treatment: • Stop protein intake • Dialysis • Increase ammonia excretion: Na benzoate, Na phenylbutyrate, L-arginine, L-citrulline
Overview • Key reaction: hydrolysis of arginine Arginine + H2O ==> urea + ornithine arginase Resynthesis of Arginine
Blood Urea Nitrogen • Normal range: 7-18 mg/dL • Elevated in amino acid catabolism • Glutamate N-acetylglutamate CPS-1 activation • Elevated in renal insufficiency • Decreased in hepatic failure
THE UREA CYCLE Urea cycle -a cyclic pathway of urea synthesis first postulated by H.Krebs • The sources of nitrogen atoms in urea molecule: • aspartate; • NH4+. • Carbon atom comes from CO2.
O O H2N-C-O-P-O- O- NH3+ NH3+ O H2N-C-NH-CH2CH2CH2CH-CO2- H2N-CH2CH2CH2CH-CO2- O H2N-C-NH2 CO2- NH3+ NH3+ -O2C-CH2CH-NH3+ CO2- NH2+ -O2C-CH2CH-NH-C-NH-CH2CH2CH2CH-CO2- NH2+ H2N-C-NH-CH2CH2CH2CH-CO2- The urea cycle HCO3- 2 ATP 2 ADP + Pi mitochondria carbamoyl phosphate NH4+ citrulline ornithine Pi Asp cytosol ATP AMP + PPi urea H2O argininosuccinate arginine fumarate -O2C-CH=CH-CO2-
O O O O NH4+ + HCO3- H2N-C-O-P-O- H2N-C-O-P-O- carbamoyl phosphate O- O- 2 ATP 2 ADP + Pi O O (1) carbonic-phosphoric acid anhydride HCO3- HO-C-O-P-O- O- ATP ADP NH4+ (2) Pi ATP ADP O carbamate H2N-C-O- (3) Incorporation of ammonia into urea begins with formation of carbamoyl phosphate This occurs in the mitochondrial matrix. Carbamoyl-phosphate synthetase-1 catalyzes the reaction in three steps, using two molecules of ATP:
O O H2N-C-O-P-O- O- NH3+ O H2N-C-NH-CH2CH2CH2CH-CO2- +H3N-CH2CH2CH2CH-CO2- NH3+ Carbamoyl phosphate reacts with ornithine to form citrulline ornithine carbamoyl phosphate Pi + H+ citrulline This step also occurs in the mitochondrial matrix.
NH3+ O H2N-C-NH-CH2CH2CH2CH-CO2- citrulline aspartate argininosuccinate CO2- NH3+ -O2C-CH2CH-NH3+ CO2- NH2+ -O2C-CH2CH-NH-C-NH-CH2CH2CH2CH-CO2- Combination of citrulline with aspartate to form argininosuccinate is driven by breakdown of ATP to AMP ATP AMP + PPi + H2O This reaction occurs only in the cytosol, so citrulline first must leave the mitochondria. A transporter exchanges ornithine for citrulline plus a proton across the mitochondrial inner membrane.
NH3+ NH3+ CO2- NH2+ -O2C-CH2CH-NH-C-NH-CH2CH2CH2CH-CO2- NH2+ H2N-C-NH-CH2CH2CH2CH-CO2- Argininosuccinate splits into arginine and fumarate argininosuccinate -O2C-CH=CH-CO2- fumarate arginine This reaction occurs in the cytosol.
O NH3+ NH3+ H2N-C-NH2 H2N-CH2-CH2-CH2-CH-CO2- H+ ornithine urea NH2+ H2N-C-NH-CH2CH2CH2CH-CO2- Hydrolysis of arginine releases urea and regenerates ornithine arginine H2O This reaction occurs in the cytosol. To continue the cycle, ornithine must return to a mitochondrion.
O O H2N-C-O-P-O- O- NH3+ O H2N-C-NH-CH2CH2CH2CH-CO2- O H2N-C-NH2 CO2- NH3+ -O2C-CH2CH-NH3+ CO2- NH2+ -O2C-CH2CH-NH-C-NH-CH2CH2CH2CH-CO2- Formation of urea consumes 4 phosphate anhydride bonds HCO3- 2 ATP 2 ADP + Pi carbamoyl phosphate NH4+ citrulline Pi ornithine ATP 2 Pi PPi + AMP Asp H2O urea argininosuccinate
The aspartate consumed in the urea cycle can be regenerated from the fumarate that is produced 2 ATP 2 ADP + Pi HCO3-+ NH4+ carbamoyl phosphate -keto acids amino acids Pi aspartate-oxaloacetate aminotransferase ornithine citrulline oxaloacetate Urea cycle ATP urea aspartate AMP + PPi malate dehydrogenase arginine argininosuccinate NADH malate NAD+ fumarate This process also uses both cytosolic and mitochondrial enzymes H2O
NADH NAD Oxidation of malate in mitochondria generates ATP 2 e- to O2 via NADH dehydrogenase generates ~ 2.5 ATP 2 ATP 2 ADP + Pi HCO3-+ NH4+ carbamoyl phosphate oxaloacetate mitochondrion Pi glutamate aspartate malate ornithine citrulline -ketoglutarate -ketoglutarate citrulline glutamate ornithine ATP aspartate urea AMP + PPi amino acids a-ketoacids arginine argininosuccinate malate cytosol fumarate H2O NADH, NAD+ and oxaloacetate can’t cross the mitochondrial inner membrane, but there are transporters for malate, aspartate, glutamate and a-ketoglutarate.
Transport systems in the mitochondrial inner membrane exchange aspartate for glutamate and a-ketoglutarate for malate mitochondrion malate aspartate- -ketoglutarate glutamate- + H+ malate aspartate- glutamate- + H+ -ketoglutarate cytosol Because the Asp/Glu transporter also moves a proton across the membrane, it can be driven by an electrochemical potential gradient. Mutations in this transporter have been linked to autism.
2 e- to electron-transport chain mitochondrion NAD+ NADH oxaloacetate malate aspartate glutamate -ketoglutarate malate aspartate glutamate -ketoglutarate oxaloacetate NAD+ NADH cytosol glycolysis Alpha--ketoglutarate/malate and aspartate/glutamate transporters also participate in oxidation of cytosolic NADH
Well Fed State Net: 2 NH4+ + CO2 + 4 ATP urea + 4 ADP + 4 Pi
Fasted State Gluconeogenesis 2 ala + CO2 1 urea + 1 glucose
Balancing the levels of ammonia and aspartate for entry into urea cycle
carbamoyl- phosphate CO2- CO2- 2 ATP 2 ADP + Pi +H3N C H CH3CO-NH C H O O CH2 CH2 H2N-C-O-P-O- NH4+ + HCO3- CH2 CH2 O- CO2- CO2- The urea cycle is regulated in two ways 1. Allosteric activation of carbamoylphosphate synthetase-1 by N-acetylglutamate + acetyl-CoA N-acetylglutamate CoA-SH Glu In mammals, N-acetylGlu appears to play only a regulatory role. Carbamoylphosphate synthetase-1 is completely inactive in its absence. A genetic deficiency in the enzyme that forms N-acetylGlu can cause a lethal defect in the urea cycle. 2. A high-protein diet or starvation leads to increased synthesis of all five enzymes used in the urea cycle, including carbamoylphosphate synthetase-1. Expression of the enzyme that synthesizes N-acetylglutamate also increases.
The Linkage between Urea Cycle, Citric Acid Cycle and Transamination of Oxaloacetate • Fumarate formed in urea cycle enters citric acid cycle and is converted to oxaloacetate. • Fates of oxaloacetate: • transamination to aspartate, • conversion into glucose, • condensation with acetyl CoA to form citrate, • conversion into pyruvate.
Diagnostic significance of the determination of urea in urine. 25-30 g/day of urea is excreted in normal conditions. The increase of urea in urine occurs in high fever, malignant anemia, poisoning by phosphorus, intensive decomposition of protein in organism. The decrease of urea in urine occurs in liver diseases, kidney unsufficiency, acidosis. Total slide : 50
Urea Cycle Disorders • Deficiency of any of the five enzymes in the urea cycle results in the accumulation of ammonia and leads to encephalopathy. • Episodes of encephalopathy and associated systems are unpredictable and, if untreated, are lethal or produce devastating neurologic sequelae in long-term survivors. • Although these disorders do not produce liver disease, the consequences of hyperammonemia resemble those seen in patients with hepatic failure or in a transient interference with the urea cycle, as seen in some forms of organic acidemias. • Investigate for hyperammonemia in any infant or child with altered mental status
The urea cycleAsterisk = N-acetyl glutamate synthetase; 1 = carbamyl phosphate synthetase; 2 = ornithine transcarbamylase; 3 = argininosuccinate synthetase; 4 = argininosuccinate lyase; 5 = arginase
UREA CYCLE DISORDERS Disorder Deficient Enzyme Inheritance Pattern Carbamyl phosphate synthetase deficiency Carbamyl phosphate synthetase Autosomal recessive Ornithine transcarbamylase deficiency Ornithine transcarbamylase X-linked Citrullinemia Argininosuccinate synthetase Autosomal recessive Argininosuccinic aciduria Argininosuccinate lyase Autosomal recessive Argininemia Arginase Autosomal recessive
Case The patient is a full-term newborn boy from a normal vaginal delivery. The pregnancy was uncomplicated. At 36 hours the baby became lethargic, irritable, and was hyperventilating. Over the next 24 hours lethargy increased and progressed to coma requiring mechanical ventilation. Hemodialysis was started at 5 days. Patient died at one week of age. Laboratory Results At 36 hours arterial blood pH was 7.50 (7.35-7.45), carbon dioxide was 25 torr (35-45), and blood urea nitrogen was 2 mg/dl (5-20). Sepsis workup was negative. On day 5 plasma ammonium was 1800 :mol/l (<35). Plasma glutamine was 1500 :mol/l (550-650),arginine was below normal, and citrulline undetectable. Orotic acid in the urine was extremely elevated. Family History Two of the mother’s four brothers had died shortly after birth. Cause of death was given as encephalitis. Biochemical Basis of Disorder , same as.. Diagnosis: ornithine transcarbamoylase deficiency
Biochemical explanations for ornithine transcarbamoylase deficiency • Low BUN • Low blood arginine • Undetectable blood citrulline • Elevated blood ammonia • Elevated blood glutamine • Elevated orotic acid
NH4+ + Glu Gln Carbamoyl P orotic acid
NH4+ + Glu Gln Carbamoyl P orotic acid