Download

1 / 36
360 likes | 630 Views
3 . 4 Genom ų s e kvenavimas. DNR sekvenavimo metodai:. Sengerio (sukurtas 1974 m., modifikuotas 1982 m.) fermentinis metodas. V.Rančeli s ( 2000). Maksamo-Gilberto cheminis metodas. DNR sekvenavimo metodai:. V.Rančeli s ( 2000). DNR sekvenavimo metodai:.

E N D
DNR sekvenavimo metodai: Sengerio (sukurtas 1974 m., modifikuotas 1982 m.) fermentinis metodas V.Rančelis (2000)
Maksamo-Gilberto cheminis metodas DNR sekvenavimo metodai: V.Rančelis (2000)
DNR sekvenavimo metodai: automatinis metodas – tai automatizuotas F. Sengerio išrastas metodas Visi dabartiniu metu naudojami genomų sekvenavimo būdai remiasi ‘Shotgun’ automatiniu DNR sekų generavimu Chromatografo kreivės
Genomo sekvenavimo tikslas gauti pilnai iššifruotą DNR grandinėlę (A, C, G, T). Strategija Bibliotekos Sekvenavimas Grupavimas/ susiejimas Dauguma genomų gali būti sekvenuoti be didesnių problemų Sulygintos sekos apjungiamos į vientisą ištisinę seką Klonų gretinimo (hierarchiniu) būdu arba viso genomo sekvenavimas per kartą Subklonavimas; ant gerai žinomų vektorių sudaromos nedidelės “įterpimo” bibliotekos Užbaigimas Atskirų DNR fragmentų apjungimas į artimas, kaimynines sulygintų sekų grupes (angl. contig) pagal jų persidengimą Anotacija • DNR savybės (pasikartojimai/panašumai) • Genų lokalizacija • Peptidų savybės • Pirminis peptidų vaidmens ar paskirties nustatymas • Kitos reguliatorinės sritys Perdavimas naudojimui
Pagrindinės genomo sekvenavimo strategijos • Hierarchinė arba klonų gretinimo (angl. Clone by clone): • DNR skaidymas į daugybę pakankamai ilgų segmentų ir didelės • kolekcija BAC klonų sudarymas. • 2. DNR fragmentų sudėliojimas į fizinius genolapius. • 3. Minimalaus fragmentų persidengimo varianto suradimas. • 4. Kiekvieno varianto klono sekvenavimas su ‘Shotgun’. • 5. Fragmentų apjungimas kaimynystės principu į klonų gretinius (angl. contig). • 6. Visų fragmentų sujungimas į vientisą seką. • Šis būdas panaudotas mielių, kirmėlių, žmogaus, žiurkės genomų sekvenavimui. • Hierarchinės strategijos atmaina yra ‘žingsniavimas’ (angl. walking), kai nesudaromi fiziniai genolapiai: • DNR skaidymas į daugybę pakankamai ilgų segmentų ir perteklinės BAC klonų kolekcijos sudarymas. • Sekvenavimas kiekvieno klono, pirmiausia parinkus keletą kertinių klonų (angl. seed clones). • Vientisos sekos konstravimas vis pridedant sekvenuotus prie jau esamų. • Tokiu būdu sekvenuotas ryžio genomas. Viso genomo sekvenavimas iš karto (WGS), išvengiant genolapių sudarymo. Šis būdas taikytas sekvenuojant drozofilos, žmogaus, pelės, žiurkės, šuns genomus.
Genomo dydis ir sekvenavimo strategijos Genomodydis (log Mb) 4 0 1 2 3 H.sapiens (3000 Mb) D.melanogaster (170 Mb) C.elegans (100Mb) P.falciparum (30 Mb) S.cerevisiae (14 Mb) E.coli (4 Mb) Viso genomo per kartą (WGS) Hierarchinis arba klonavimo Visos chromosomos (WCS) Viso genomo per kartą ‘Shotgun’ (WGS) derinant su BAC mažo padengimo klonais (panaudotas žiurkės genomo sekvenavimui) www.sanbi.ac.za/mrc/tdr2004/Presentation/ genomics_lawson/sanbi_genome_files/
Vektoriai – struktūros, kuriose laikoma įterpta klonuota svetima DNR. Tai atliekama dviem tikslais: saugoti ir dauginti klonuotą DNR. Vektorių tipai
DNR sekvenavimas – vektoriai DNR DNR fragmentai Žinoma vieta (restriktazių pažinimo vieta) Vektorius (žiedinė plazmidės DNR) + =
Genetiniai genolapiai sudaromi pasinaudojant rekombinacijų dažniais ir santykiniais atstumais (cM), o fiziniai – pasinaudojant fiziniais klonuotų DNR sekų atstumais. • Genetiniai žymenys naudojami kaip gairės sudarant genolapius: • morfologiniai kategoriniai žymenys, pvz. žmonių hemofilija, daltonizmas, žirnelių raukšlėtumas; • Fiziniai žymenys - RFLP, CAPS, VNTR, STS.
http://www.ornl.gov/sci/techresources/Human_Genome/home.shtmlhttp://www.ornl.gov/sci/techresources/Human_Genome/home.shtml
Komplementaraus jungimosi būdu gaunamas tikslus molekulinių žymenų išsidėstymas chromosomoje Detalus genolapio STS (angl. sequence-tagged-site) pagrindu sudarymas remiasi klonuotų BAC fragmentų kartografavimu, persidengimo principu (angl. contig).
Sekvenavimo strategijų privalumai ir trūkumai • Hierarchinis sekvenavimas • Privalumai: • Lengvesnis apjungimas sekvenuotų • fragmentų į klonų gretinius • - Reikalauja mažiau kompiuterinių • resursų • Yra patikimesnis • Trūkumai: • Reikalingi fiziniai genolapiai ir klonų • bibliotekos • Daug perteklinio padengimo (angl. redundant sequencing) • Techniškai sudėtingas ir pakankamai brangus • Viso genomo sekvenavimas per kartą • Privalumai: • Nereikia genolapių • Mažiau perteklinio padengimo • - Reikalauja daugiau kompiuterinių • resursų • Yra pigesnis • Trūkumai: • Sudėtingas sekvenuotų fragmentų apjungimas į klonų gretinius, t.y. eksperimentiškai sudėtingesnis • Reikalauja daugiau kompiuterinių • resursų • - Nėra patikimas
Eukariotų genomų anotacija ab initio genų suradimas Genominė DNR transkripcija Pirminė RNR RNR procesingas Brandi mRNR Gm3 AAAAAAA Lyginamasis genų išaiškinimas transliacija Susidarantis polipeptidas susisukimas Aktyvus enzimas Funkcinė identifikacija Funkcija Reaktantas A Produktas B Genomo anotacija ar išaiškinimas – tai genus atitinkančių DNR sekų identifikavimas, siekiant atsakyti į klausimus: kiek genų yra ir kuriose genomo vietose, kokius baltymus jie koduoja, kokie yra reguliaciniai mechanizmai ir sąveikos schemos.
Struktūrinė anotacija susijusi genų vietos suradimu, homologinių su kitais genomais, cDNA sekomis ir baltymų sekomis DNR grandinės vietų paieška, o taip pat transkripciją reguliuojančių elementų identifikavimu. Funkcinė anotacija susijusi su baltymų molekuline funkcija, jų dalyvavimu apykaitos ir reguliatorinėje veikloje. E.coli genomo vietos fragmentas
Egzonas 3 Egzonas 1 Egzonas 2 Intronas 1 Intronas 2 5’ 3’ Stop kodonas TAG/TGA/TAA Start kodonas ATG Susijungimovietos Ab initio genų metodai paremti specifinių vietų sekose paieška, tokių kaip start ir stop kodonai, ir ribosominės kilpos. Jei nukleotidai sekoje išsidėstę atsitiktinai (tai būdinga introninėms geno dalims, tarpgeninei DNR), maždaug kas dvidešimtas nukleotidų trejetas esti atsitiktinis stop kodonas. Palyginti ilgas DNR fragmentas, kuriame nėra stop kodonų, vadinamas atviru skaitymo rėmeliu (ORF). GenScan, Genie, GeneID kompiuterinės programos yra naudojamos tokio pobūdžio analizei.
Kiti genų identifikavimo metodai grindžiami homologija su jau žinomais genais (naudojama GenomeScan kompiuterinė programa). Tokie yra, pvz.: - zooblotingas, kai hibridizuojant naujai nustatyto žmogaus geno DNR sekas su žinomais kitų rūšių (beždžionių, galvijų, pelių, paukščių) genais, galima identifikuoti tų genų analogus žmogaus genome (GeneWise, Procrustes ir kt. kompiuterinės programos); - CpG salelės, kurių buvimas nustatytose DNR sekose padeda rasti visą geną. Šios salelės labiau būdingos bendriniams (angl. housekeeping) genams, pvz., tokiems, kurie koduoja ląstelės energetikai būtinus baltymus ir kt. (Rosseta, SGP1 kompiuterinės programos); - egzono įterpimas (angl. exon trapping) paremtas žiniomis apie tam tikras nukleotidų sekas eukariotų genuose žyminčias introno pradžią ir pabaigą. Jei gautos mDNR ilgis pasikeičia, vadinasi svetimos DNR egzonas buvo atpažintas ir prijungtas (CEM kompiuterinė programa).
Genai yra identifikuojami pasinaudojant ekspresuotų sekų žymekliais (EST). Idėja grindžiama tuo, kad identiškos sekos atlieka panašų vaidmenį ir kituose genomuose. Specialios kompiuterinės programos padeda identifikuoti genus pagal nukleotidų sekų išreikštumą (BLASTN, FASTA, TBLASTN). Naujesnės kompiuterinės programos, tokios kaip SGP-2, TwinScan, SLAM, DoubleScan yra sukonstruotos panašumo principu ir naudojamos homologiniams genomų lyginimams. • Iškylantys sunkumai: • - sunku tiksliai iškart nustatyti genų skaičių • visas genomas sekvenuotas su kai kuriomis pasitaikančiomis klaidomis • mažus genus yra sunku identifikuoti • kai kurie genai retai pasireiškia ir neturi būdingos kodonų struktūros, todėl juos sunku aptikti • genų funkcijos daugumoje yra nežinomos
Naujai įsitvirtinantys sekvenavimo metodai Hibridinis sekvenavimas (SBH) Daugybinis spektrometrinis sekvenavimas Tiesioginė atskiros DNR molekulės vizualizacija naudojant atominę mikroskopiją (AFM) Atskiros molekulės sekvenavimas Atskiro nukleotido metodas Geno ekspresijos ląstelėje nustatymo metodas Sekvenavimas panaudojant nanoporą www.ist.temple.edu/~vucetic/ cis595spring2003/
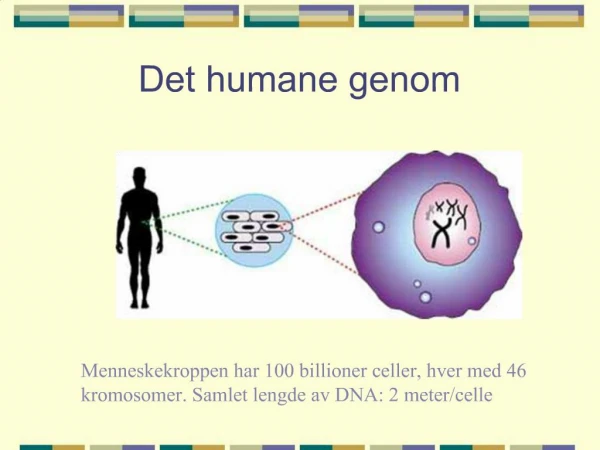
Žmogaus genomas 1 2 3 X 6 16 7 mitochondrija 11 4 19 20 8 5 9 10 17 18 12 13 22 15 21 14 Y .016 45 66 72 48 51 104 3.2*109 bp 86 88 100 107 163 118 148 143 142 140 176 163 148 221 279 198 197 Myoglobinas a globinas 251 *5.000 b-globinas (11 chromosoma) 6*104 bp *20 Egzonas 1 Egzonas 2 Egzonas 3 3*103 bp 5’ UTR 3’ UTR *103 DNR: ATTGCCATGTCGATAATTGGACTATTTGGA 30 bp Baltymas: http://www.sanger.ac.uk/HGP/ aa aa aa aa aa aa aa aa aa aa
Žmogaus genomas • Genai sudaro ~ 25% viso genomo • Egzonai užima tik 1% • Vidutinis žmogaus genas: • 27kb ilgio ir koduojančia seka sudarančia 1,340 bp • Tik 5% genų atitinka koduojančias sekas (genai skiriasi pagal intronų skaičių) www.gmu.edu/departments/ biology/568-0304.ppt
Egzonai: baltymuskoduojantys irnetransliuojamos sritys (UTR) 1 to 178 egzonųgali turėti genas (vidurkis 8.8) 8 bp to 17 kb gali sudaryti egzoną (vidurkis 145 bp) • Intronai: nekoduojančios DNR sekos vidutiniškai 1 kb – 50 kb sudaro introną • Genų dydis: Didžiausias – 2.4 Mb (Distrofinas). Vidurkis – 27 kb. Eukariotų genomai turi santykinai mažai koduojančių sričių Eukariotų genomus didžiąja dalimi sudaro kartotinės sekos Eukariotų genai yra grupuojami blokuose tarp kartotinių sekų Dėl minėtų priežasčių yra reikalingi paprastesni “modeliniai” genomai
http://www.genomesonline.org/ •Publikuotipilni genomai:359 •Prokariotųvykstantysgenomų dekodavimai:944 •Eukariotųvykstantysgenomų dekodavimai:599 •Viso: 1902 2006 metų (pirmo ketvirčio) duomenys
Augalų genomai skiriasi dydžiu, ploidiškumu ir chromosomų skaičiumi Arabidopsis 125.000 Kb Bananas (Musa) 873.000 Kb Lilium 50.000.000 Kb http://www.redbio.org/portal/encuentros/enc_2001/conferencias/C-04/
Ląstelių tipų ir morfologinis kompleksiškumas Genomų dydis nėra proporcingas organizmų kompleksiškumui
pg=1012g Eriksson and Ekberg (2001)
Kokios galimospriežastys genomų dydžio skirtumų tarp baltažiedžio vairenio (Arabidopsis) ir daugumos spygliuočių medžių rūšių? - Daugiau resursų reikalaujančios DNR sintezė, matyt, nėra didelis kliuvinys - Svarbu pažymėti, kad didelis genomas koreliuoja su dideliu ląstelės branduoliu, o branduolio dydis savo ruožtu su lėtesne mitozės ir mejozės dalijimosi eiga - Paminėta ypatybė nėra svarbi medžių rūšių išlikimui. Medžiai ir žolės augančios šiaurinėse platumose pasižymi labai nedideliu DNR turiniu. Tikėtina, kad tą lemia trumpas vegetacijos sezonas, nes augalams reikia praeiti keletą vystymosi stadijų - Arabidopsio gyvenimo ciklas labai trumpas 2-3 savaitės nuo sėklos iki sėklos. Tik mažas augalo genomas gali leisti tokį spartų ląstelių dalijimąsi - Pozityvi didelio genomo selekcija vyksta daugelyje spygliuočių medžių rūšių. Paprastai šios rūšys vietoj vandens indų turi tracheides. Yra nustatytas priežastinis teigiamas ryšys tarp tracheides produkuojančių kambio ląstelių ir branduolio DNR dydžio. 18 Š.Amerikos pušų rūšių tyrimai parodė, kad rūšys prisitaikę gyventi nepalankiomis sąlygomis (pusdykumėse) turi didesnį genomą nei augančios optimaliose sąlygose. Eriksson and Ekberg (2001)
Literatūros sąrašas Brown G.R., Gill G.P., Kuntz R.J., Langley C.H. and Neale D.B. 2004. Nucleotide diversity and linkage disequilibrium in loblolly pine. PNAS 101 (42): 15255–15260. BrownGR., KadelEE. III, BassoniDL., KiehneKL., TemesgenB., BuijtenenJP. van, SewellMM., MarshallKA., NealeDB., vanBuijtenenJPAD 2001. Anchored reference loci in loblolly pine (Pinus taeda L.) for integrating pine genomics.Genetics, 159 (2): 799-809. Eriksson G. and Ekberg I. 2001. An introduction to forest genetics. SLU Repro, Uppsala. Pp. 166. Neale DB. and Savolainen O. 2004. Association genetics of complex traits in conifers. Trends in Plant Science 9: 325-330. Pavy N., Paule Ch., Parsons L., Crow J.A., Morency M-J., Cooke J., Johnson J.E., Noumen E., Guillet-Claude C., Butterfield Y., Barber S., Yang G., Liu J., Stott J., Kirkpatrick R., Siddiqui A., Holt R., Marra M., Seguin A., Retzel E., Bousquet J. and MacKay J. 2005. Generation, annotation, analysis and database integration of 16,500 white spruce EST clusters. BMC Genomics 6 (144): 1-19. Rančelis V. 2000. Genetika. Lietuvos Mokslų Akademijos leidykla, Vilnius. Pp. 662. Steane D. 2005. Complete Nucleotide Sequence of the Chloroplast Genome from the Tasmanian Blue Gum, Eucalyptus globulus (Myrtaceae). DNA Research 12: 215-220.