Download
1 / 84
890 likes | 1.05k Views
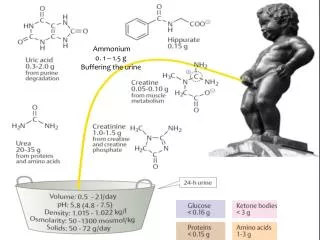
Ammonium 0. 1 – 1.5 g Buffering the urine . AMINO ACID METABOLISM A mino acids are required for the synthesis of proteins, peptides, nucleotides, neurotransmitters, other amino acids

E N D
Ammonium 0. 1 – 1.5 g Buffering the urine
AMINO ACID METABOLISM • Amino acids are required for the synthesis of proteins, peptides, nucleotides, neurotransmitters, other amino acids • Free amino acids can be provided to cells either from the digestion of dietary proteins or the degradation of defective or aged cellular proteins • Amino acids are catabolized into components that can directly join energy production pathways or be changed to glucose, fatty acids or ketone bodies; this happens: • when the amount of amino acids obtained from digestion and degradation is more than what is needed for biosynthesis • during starvation or uncontrolled diabetes mellitus • The catabolism of amino acids produces: • amino group – removed as urea • carbon skeleton – seven types of intermediates
The digestion of proteins • The digestion of proteins begins in the stomach and continues in the small intestine • Please refer to the notes on enzymes for the types, activation and specificity of the proteases involved in the digestion • Rennin (chymosin) is important in infants because it breaks a specific peptide bond in casein (a milk protein) curdling the milk and increasing transit time in the stomach • Proteases can be endopeptidases or exopeptidases • The endopeptidases are specific for different types of peptide bonds and produce fragments of varying sizes • Exopeptidases take over the job: • carboxypeptidase A and B –produced by the pancreas and cleave at the C-terminal • brush border aminopeptidases– act on the N-terminal of oligopeptides yielding free amino acids and di- and tripeptides
Digestive enzymes themselves are digested and contribute to the amino acid pool • Free amino acids are transported into intestinal epithelial cells by Na+ - dependent secondary transport • Di- and tripeptides enter the epithelial cells through symport with H+ • theH+ gradient is maintained by the Na+ - H+ exchanger • peptidases in the epithelial cells change the di- and tri- peptides into free amino acids • Amino acids enter the portal vein by facilitated transport • Many cells use Na+ - dependent secondary transport and to some extent facilitated transport in order to absorb amino acids • premature activation of zymogens inside the pancreas results in acute pancreatitis
Protein turnover • The degradation and resynthesis of proteins • The half-lives of eukaroytic proteins may vary from 30 seconds to many days • ornithinedecarboxylase – approx. 11 minutes; hemoglobin – lifespan of red blood cells; crystallin (lens protein) – life span of the organism • Rapidly degraded proteins include those proteins that are defective due to wrong insertion of amino acids or damage accumulated during normal functioning, regulatory enzymes, • A protein's half-life correlates with its N-terminal residue • Proteins with N-terminal Met, Ser, Ala, Thr, Val or Gly have half lives greater than 20 hours • Proteins with N-terminal Phe, Leu, Asp, Lys or Arg have half lives of 3 min or less
PEST proteins having domains rich in Pro (P), Glu (E), Ser (S) and Thr (T) are more rapidly degraded than other proteins • There are two ways of intracellular degradation of proteins • lysosomal degradation - of endocytosed proteins or proteins undergoing autophagy • In autophagy, part of the cytoplasm may become surrounded by two concentric membranes • Fusion of the outer membrane of this autophagosome with a lysosomal vesicle results in the degradation of enclosed cytoplasmic structures and macromolecules • the enzymes • responsible for the • degradation are • cathepsins
ATP- dependent cytosolic degradation • The ubiquitin – proteasome pathway • Ubiquitin is a small protein having 76 amino acids only • It is present in all eukaryotes (hence the name) and its amino acid sequence is highly-conserved • Ubiquitin marks proteins for death • the carboxyl terminal of a ubiquitin forms an isopeptide bond with the ε-amino group of a lysine residue of a protein to be destroyed • Three enzymes, are involved in the attachment of ubiquitin: • Initially the terminal carboxyl group of ubiquitin is joined in a thioester bond to a cysteine residue on Ubiquitin-Activating Enzyme (E1) ; this is an ATP-dependent process
The ubiquitin is then transferred to a sulfhydryl group on a Ubiquitin-Conjugating Enzyme (E2) • Ubiquitin-Protein Ligase (E3) then promotes the transfer of ubiquitin from E2 to the ε-amino group of a Lys residue of a protein recognized by that E3 • The substrate-specificity of this system comes from the various combinations of the types of E2 and E3 • More ubiquitins are added to form a polyubiquitin chain • The terminal carboxyl of each ubiquitin is linked to the ε-amino group of a lysine residue (Lys 29 or Lys 48) of the adjacent ubiquitin • A chain of 4 or more ubiquitins targets proteins for degradation in proteasomes
The proteasome is a complex of multiple proteases • Constitutes nearly 1 % of cellular protein • It contains two main types of subcomplexes: a barrel-like core particle (20S) and regulatory 19S particles on both ends of the barrel • the catalytic core particle and the regulatory particles make up the functional 26S proteasome • The 19 S particles may unfold proteins and translocate the unfolded proteins into the 20 S particle; energy from ATP is consumed in the process • The 19 S particles also cleave isopeptide bonds and free ubiquitin; ubiquitin is recycled • The protein is degraded by the 20 S particle and free amino acids are released into the cellular space
Proteasomal degradation of particular proteins is an essential mechanism by which cellular processes are regulated, such as cell division, apoptosis, differentiation and development • progression through the cell cycle is controlled in part through regulated degradation of proteins called cyclins that activate cyclin-dependent kinases • Inability to degrade proteins that activate cell division (or rapid degradation of those proteins that suppress tumor formation) can lead to cancer • Diseases like Alzheimer's, Parkinson’s , type II diabetes,… are associated with the deposition on tissues of non-degradable protein aggregates known as amyloid
The catabolism of amino acids • The fate of amino acid nitrogen • α-amino groups are removed from amino acids mostly through transaminationreactions • the amino acid becomes a keto acid when it donates the amino group to α-ketoglutarate (changing it to glutamate) • All amino acids except lysine and threonine undergo transamination reactions • The enzymes involved are known as transaminases or aminotransferases • Pyridoxal phosphate (PLP) is the cofactor • The glutamate thus derived collects the amino groups and gives them off for biosynthesis or excretion
Free ammonium can be released in different ways: • The oxidative deamination of glutamate by glutamate dehydrogenase - the only enzyme that can use NAD+ or NADP+ as electron acceptor • The reaction is reversible and takes place in the mitochondria • Serine and threoninedehydratases release NH4+; histidine can be directly deaminated to give NH4+ • Intestinal bacteria produce NH4+ from amino acids or urea; the ammonia enters the portal vein • Glutamine and asparagine lose their side chain amino groups through deamidation • The purine nucleotide cycle in the brain – aspartate is used as a substrate and fumarate and ammonium are released
The urea cycle • Nitrogen balance is the difference between the amount of nitrogen consumed and excreted per day • Positive nitrogen balance in growing individuals • Negative nitrogen balance during protein deficiency or starvation • In healthy adults, the amount of nitrogen consumed and excreted is approximately equal • Nitrogen is excreted in the form of urea, uric acid, ammonium, creatinine, hippurate, creatine • Humans are ureotelic (excrete excess nitrogen mainly in the form of urea) • ammonotelic (ammonia); uricotelic (uric acid) • Ammonium made available in the liver from different sources enters the urea cycle
Since the enzymes of the urea cycle are present in the liver only, amino groups from other tissues should be transported to the liver • Two mechanisms of transport: • the skeletal muscles export alaninesynthesized from the transamination of pyruvate (glucose catabolism) by glutamate • the amino group donated by glutamate was obtained from the breakdown of amino acids in the muscle
Glutamate can accept another amino group through an ATP-dependent reaction catalyzed by glutamine synthetase • Glutamine is used by most tissues to transport ammonium • Glutamine travels to the liver, kidneys and the intestine and is deamidated • the ammonium released by the deamidation is used as a buffer (in the kidneys) or enters the urea cycle in the liver; glutamine can be used as an energy source by the intestine
The two nitrogen atoms of urea enter the urea cycle as NH4+ and as the amino N of aspartate • The synthesis of carbamoyl phosphate • The NH4+ and HCO3- (carbonyl C) that will be part of urea are incorporated first into carbamoyl phosphate • The cleavage of 2 ATP molecules is needed to form the high energy carbamoyl phosphate • Carbamoyl phosphate synthetase (CPS I) is a mitochondrial enzyme; the cytosolicisozyme is involved in pyrimidine synthesis • CPS I has an absolute requirement for the allosteric activator N-acetylglutamate • This derivative of glutamate is synthesized from acetyl-CoA and glutamate when cellular glutamate is high, signaling an excess of free amino acids due to protein breakdown or dietary intake
The formation of citrulline • Carbamoyl phosphate reacts with ornitihine to give citrulline; catalyzed by ornithinetranscarbamoylase • The entry of the second N • Citrulline leaves the mitochondria in exchange for the entry of ornithine from the cytosol • Citrulline reacts with aspartate producing argininosuccinate • Argininosuccinate synthetase requires the splitting of ATP to AMP and PPi
AMP+ PPi ATP argininosuccinate synthetase + Pi + ornithine trans- carbamoylase
The formation of arginine • Argininosuccinate lyase produces arginine and fumarate • The arginine produced by the urea cycle is enough for adults • The carbons of fumarate are those that were obtained from aspartate • Fumarate be changed to oxaloacetate by enzymes of the citric acid cycle • The oxaloacetate will receive an amino group from glutamate and be changed to aspartate; aspartate reenters the urea cycle • The TCA and urea cycles constitute a bicycle: TheKrebs bicycle • The production of urea and the regeneration of ornithine • The action of arginase produces urea and ornithine • Urea travels to the kidneys and excreted through the urine
The Krebs bicycle • The stocihiometry of the urea cycle • NH4++ CO2 + 3 ATP+ 2H2O→urea + fumarate + 2 ADP + AMP+ 4Pi
In addition to the allosteric effects of N-acetylglutamate, the synthesis of the enzymes of the urea cycle can be induced during periods of increased metabolism – protein rich diet or prolonged fasting • Urea cycle abnormalities • Hereditary deficiency in any one of the urea cycle enzymes or liver cirrhosis lead to the increase in the blood of ammonia (hyperammonemia) or urea cycle intermediates • The total lack of any urea cycle enzyme is lethal • Elevated ammonia is toxic, especially to the brain • Why is ammonia toxic to the brain? Hypotheses: • High ammonia levels would drive glutamine synthetase; this would deplete glutamate – a neurotransmitter and precursor for the synthesis of GABA • Glutamine may exert osmotic effects leading to the swelling of the brain
Depletion of glutamate and high ammonia levels would drive the glutamate dehydrogenase reaction in the reverse direction; the resulting depletion of α-ketoglutarate, inhibits the production of energy • The treatment of urea cycle defects • limiting protein intake to the amount barely adequate to supply amino acids for growth, while adding to the diet the α-keto acids of essential amino acids • liver transplantation; gene therapy has also been tried • If the defect occurs after the synthesis of argininosuccinate, argininosuccinate can be used as a carrier for the removal of nitrogen (because it has incorporated both amino groups) • the problem in this situation would be one of regenerating ornithine • If arginase is not deficient, the intake of high amounts of arginine would provide ornithine
If the defect occurs at a point before the synthesis of argininosuccinate, substances are used that form conjugates with amino acids and are excreted in the urine • The body has to use ammonia to replace the excreted amino acids; ammonia levels decrease • Drugs:Benzoic acid - reacts with glycine to give hippurate • Phenylbutyrate - first changed to phenylacetate and then reacts with glutamine to produce phenylacetylglutamine • The most common defect is in ornithinetranscarbamoylase • In the rare cases of arginase deficiency, arginine should be excluded from the diet • Deficiency of N-acetyl glutamate can be corrected by administering an analog, carbamoyl glutamate
The fate of the carbon skeleton of amino acids • The deamination of most amino acids yields α- keto acids that, directly or via additional reactions, feed into major metabolic pathways • Amino acids are grouped into two classes based on whether or not their carbon skeletons can be converted to glucose: glucogenicorketogenic • Carbon skeletons of glucogenic amino acids are degraded to pyruvate or a 4-C or 5-C intermediates of the Krebs cycle • Glucogenic amino acids are a major carbon source for gluconeogenesis when glucose levels are low • They can also be catabolized for energy production or converted to glycogen or fatty acids for energy storage • Carbon skeletons of ketogenic amino acids are degraded to acetyl-CoA or acetoacetate
Carbon skeletons of ketogenic amino acids can be catabolized for energy or converted to ketone bodies or fatty acids • leucine and lysine are strictly ketogenic • isoleucine, threonine, phenylalanine, tyrosine and tryptophan and both glucogenic and ketogenic • The remaining thirteen amino acids are glucogenic • amino acids producing pyruvate can be considered ketogenic because pyruvate can be changed to acetyl-CoA • One carbon transfer is a common theme in amino acid metabolism • Three cofactors are used to transfer different one carbon groups between intermediates • Tetrahydrofolate (THF); S-adenosylmethionine/SAM/ado-Met; Vitamin B 12 (5’-deoxyadensoyl/methyl cobalamin) • Biotin is involved in the transfer of the most oxidized form of carbon – CO2
Tetrahydrofolate • Folic acid/folate/folacin is composed of a pteridine nucleus, para amino benzoic acid and one or more glutamic acid residues • Once folic acid is absorbed by the intestine, it is converted to the biologically active form, tetrahydrofolate , by dihydrofolate reductase • only one glutamic acid remains; 4 hydrogens added • THF travels to the liver and glutamic acid residues are added • Most of the THF is released into the bile and recirculates just like the bile acids • The carbon units carried by THF are attached to N5and/or N10 of the pteridine ring • One carbon units carried by THF are: • Most reduced: - CH3 (methyl) • Intermediate: - CH2 - (methylene) • Most oxidized: - CHO (formyl) • - CHNH (formimino) • - CH = (methenyl)
The collection of one carbon units attached to THF is known as the one – carbon pool • While they are still attached to THF, the one carbon units can be oxidized or reduced • The main source of carbon units for THF is the carbon removed during the conversion of serine to glycine producing N5, N10-methylene THF • Although THF can carry a methyl group at N5, the transfer potential of the methyl group is insufficient for most biosynthetic reactions; anothercofactor is used as a carrier of methyl groups • S-adenosylmethionine • It contains an activated methyl thioether group; donates methyl groups to oxygen or nitrogen • Its synthesis to be discussed under the synthesis of cysteine
Vitamin B 12 • The unmodified form of vitamin B 12 is known as cobalamin • It has a corrin ring similar to porphyrin • but unlike porphyrin two of its pyrrole rings are joined directly (no bridges); cobalt takes the place of iron • Cobalamin in the diet can be found in a free form or bound with proteins • Free cobalamin is then bound by salivary or gastric secretions known as haptocorrins; protein-bound cobalamin is first freed of the proteins and then haptocorins bind it • Haptocorin bound cobalamin is changed to free cobalamin in the intestine • Cobalamin is then bound by intrinsic factor which assists in the absorption into the intestine; travels to the liver bound with transcobalamin II
The transport of vitamin B 12 • Vitamin B 12 is involved in two reactions in the body: • The intramolecular rearrangement of a proton during the formation of succinyl-CoA from propionyl-CoA • the coenzyme form of vitamin B 12 used in this case is 5’- deoxy -adenosylcobalamin – 5’- deoxyadenosine attached to cobalt • The regeneration of methionine (to be discussed) • methyl is attached to cobalt to give methyl cobalamin
Vitamin B 12 X = 5’- deoxyadenosine or methyl
Essential and non-essential amino acids • Eleven amino acids are considered to be non-essential because they can be synthesized in the body • arginine, cysteine, tyrosine andhistidine are conditionally essential • Arginine and histidine needed in the diet of children and pregnant women; in adults arginine from the urea cycle is enough and histidine is effectively recycled • tyrosine and cysteine are synthesized from the essential amino acids phenylalanine and methionine, respectively; if these precursors are absent in the diet, then the products become essential • The remaining essential amino acids are lysine, leucine, isoleucine, valine, tryptophan, threonine and histidine • Except tyrosine and cysteine, essential amino acids can be synthesized from glucose and ammonia (or another amino acid)
Amino acids related with intermediates of glycolysis • In the synthesis of serine, 3-phosphoglycerate is sequentially oxidized, transaminated and dephosphorylated • In addition to the serine dehydratase reaction, serine can be changed to pyruvate through transamination followed by reduction and phosphorylation to give PEP (PEP then changed to pyruvate) • The main pathway of glycine synthesis is from serine – serine hydroxymethyltransferase catalyzes the reaction which involves PLP and N5, N10methylene THF • Glycine can be degraded by changing it to serine and then to pyruvate • A second way for the degradation of glycine is the production of glyoxylate by D-amino acid oxidase • D-amino oxidase is thought to act in the detoxification of D-amino acids from bacteria or cooked foodstuffs
Glyoxylate can react with α- ketglutarate and be directed to energy production • It can also be oxidized to oxalate by hepatic lactate dehydrogenase • This oxalate, along with the oxalate obtained from the diet, contribute to the formation of kidney stones • 3/4th of kidney stones is composed of calcium oxalate • The third and major approach to degrading glycine is by glycine cleavage enzyme • Glycine degraded to NH4+, CO2 and –CH2– (carried by THF) • If glycine cleavage enzyme is deficient, non-ketotichyperglycinemiaresults; mental retardation and early death probably due to the increased inhibitory effects of glycine on the nervous system
The carbon skeleton and the amino group of cysteine are derived from serine; the sulfur is transferred from methionine • The sulfur of methionine attacks the 5’ carbon of the ribose of ATP releasing all the three phosphates in the process; SAM (Ado met) is the product • SAM donates CH3 and becomes S-adenosylhomocysteine • The hydrolytic removal of the adenosine gives homocysteine • Serine reacts with homocysteine to give cystathionine – catalyzed by cystathionine-β–synthase • Cystathionine is cleaved by cystathionaseto give cysteine and α- ketobutyrate(which will be changed to propionyl-CoA and then succinyl-CoA) • Homocysteine can be changed to methionine: N5-methyl THF donates CH3 to cobalamin to give methyl cobalamin; methyl cobalamin donates the CH3 to homocysteine and methionine will be formed