Translational Bioinformatics
Translational Bioinformatics. Haiyan Huang May 12, 2010. Credits of some slides: Atul Butte, Stanford U Russ Altman, Stanford U . Central Dogma of Biology. Background: bioinformatics in the post-genomic era. DNA level gene annotation, functional elements identification mRNA level


Translational Bioinformatics
E N D
Presentation Transcript
Translational Bioinformatics Haiyan Huang May 12, 2010 Credits of some slides: Atul Butte, Stanford U Russ Altman, Stanford U
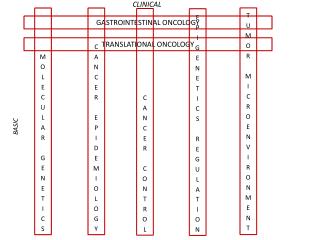
Central Dogma of Biology Background: bioinformatics in the post-genomic era • DNA level • gene annotation, functional elements identification • mRNA level • transcription regulation, gene co-expression, pathways • Protein level • protein structure/function; protein-protein interaction • System Biology • complex interactions in biological system • Translational bioinformatics • integrative information retrieval for aiding disease diagnosis
What is Translational Bioinformatics? • (by Russ Altman, MD, PhD) Using the toolkit of bioinformatics to understand the relationship of molecular information to diseases and symptoms, in order improve diagnosis, prognosis, and therapy.
What is translational bioinformatics? (by Atul Butte, MD, PhD) • Translational bioinformatics • Development of analytic, storage, and interpretive methods • Optimize the transformation of increasingly voluminous genomic and biological data into diagnostics and therapeutics for the clinician (Research on the development of novel techniques for the integration of biological and clinical data) • End product of translational bioinformatics • Newly found knowledge from these integrative efforts that can be disseminated to a variety of stakeholders, including biomedical scientists, clinicians, and patients
Why translational bioinformatics? • There is an increasing call for translational medicine: Universities, Congress, NIH, and elsewhere: “What did we get for our money?” • Incredible amounts of publicly-available data • GenBank: Hundreds of organisms have been completely sequenced • GEO, ArrayExpress has numerous samples from thousands of experiments • NCBI dbGAP for the interaction of genotype and phenotype. Such studies include genome-wide association studies, medical sequencing, association between genotype and non-clinical traits, etc
Translational Bioinformatics Tasks • Using high-throughput genomic measurements for improved diagnosis/prognosis/therapy • New classifications of disease based on molecular markers • Identify new drug targets based on molecular profiling of disease • Understanding disease pathology and genetic pathways in complex multigenic disorders • Create systems for physician decision support using genetic information
Transforming Public Gene Expression Repositories into a Disease Diagnosis Database Reference: Huang H, Liu C, Zhou XJ (2010). Bayesian Approach to Transforming Public Gene Expression Repositories into Disease Diagnosis Databases. Proc Natl Acad Sci.USA. 2010 Apr 13;107(15):6823-8.
Example project in translational bioinformatics Transforming public expressionrepositories into a disease diagnosis database • The public microarray data increases by 1.5 folds per year • NCBI Gene Expression Omnibus (GEO): > 330,000 experiments • EBI Array Express: > 115000 experiments • Largest database systematically documenting the genome-widemolecular basis of diseases • heart disease, mental illness,infectious disease, and a wide variety of cancers. Joint work with X.J. Zhou (USC), J.C. Liu (USC)
Example project in translational bioinformatics Transforming public expressionrepositories into a disease diagnosis database • The public microarray data increases by 1.5 folds per year • NCBI Gene Expression Omnibus (GEO): > 270000 experiments • EBI Array Express: > 115000 experiments • An unprecedented opportunity to study human diseases • Expression-based-diagnosiswould be particularly useful when the potential disease is notobvious or when the disease lacks biochemical diagnostic tests Joint work with X.J. Zhou (USC), J.C. Liu (USC)
Our goal: turning the NCBI GEO into an automateddisease diagnosis system • Thus far, no effective method is available for this purpose. Existing approaches have been • of limited scale, i.e., within single laboratories, • targeting specifictypes of disease, • lacking the integration of the heterogeneous datasets (i.e., from different experimental sources, with diverse phenotypes, containing the information in different formats ). Joint work with X.J. Zhou (USC), J.C. Liu (USC)
Phenotype Concepts (e.g. diseases, perturbations, tissues ) in Unified Medical Language System (UMLS) Integrating publicrepositories involves“combining the expression data and the text information” Expression data Microarray Data Phenotype information
Three challenges towards our goal • The gene expression data from different laboratories cannot be compared directly due toplatform differences and systematic variation • Thedisease and phenotype annotations of datasets are heterogeneous andembedded in text, and thus not in a workable format • Thedisease diagnosis approach must robustly characterize a queryexpression profile by jointly utilizing the large amount of noisy genomicand phenotypic data Joint work with X.J. Zhou (USC), J.C. Liu (USC)
Collecting microarray data sets … Adenocarcinoma Asthma Glaucoma • We initially collected 421 human microarrray datasets of the platform U95, U133 and U133 plus 2 from NCBI GEO. • These three major platforms share a large number of overlapping genes (8,358 genes) • We further selected100 datasets (9169 arrays) having subset types: • disease state • normal, control, non-tumor, healthy, or benign • This serves as the initial database to test our framework Joint work with X.J. Zhou (USC), J.C. Liu (USC)
Method highlights • Data Preparation • Standardizing the expression data to remove cross-lab and cross-platform incompatibilities (challenge 1). • Phenotypically annotating the collected human microarray experiments by Unified Language Medical System (UMLS) (challenge 2). • Bayesian disease inference (challenge 3) • Bayesian belief network for refining the results (challenge 3) Joint work with X.J. Zhou (USC), J.C. Liu (USC)
Log-rank-ratio Disease profile Control profile standardized profile Method highlights • Data Preparation • Standardizing the expression data to remove cross-lab and cross-platform incompatibilities (challenge 1). • Phenotypically annotating the collected human microarray experiments by Unified Language Medical System (UMLS) (challenge 2). • Bayesian disease inference (challenge 3) • Bayesian belief network for refining the results (challenge 3) Joint work with X.J. Zhou (USC), J.C. Liu (USC)
Log-rank-ratio Disease profile Control profile standardized profile Method highlights • Data Preparation • Standardizing the expression data to remove cross-lab and cross-platform incompatibilities (challenge 1). • Phenotypically annotating the collected human microarray experiments by Unified Language Medical System (UMLS) (challenge 2). • Bayesian disease inference (challenge 3) • Bayesian belief network for refining the results (challenge 3) Are the cross-dataset comparisons by standardized profiles consistent with the phenotype annotations? Joint work with X.J. Zhou (USC), J.C. Liu (USC)
Data Standardization: an example Figure.(a) Scatterplot of expression profiles on the samesamples GSM21236 (GDS817: Breast Cancer cells MDA-MB-436) versus GSM21242 (GDS820: Breast Cancer cells MDA-MB-436); (b) Scatterplot of expression rank profiles of the same samples as in (a); (c) Scatterplot of expression ratio profiles GSM21240 / GSM21236 (from GDS817) versus GSM21246 / GSM21242 (from GDS820) in log scale; (d) Scatterplot of expression rank ratio profiles of the same sample pairs as in (c) in log scale. It is obvious that log rank ratio profiles of the same sample pairs are comparable across platforms. Figure 1.(a) Scatterplot of original expression profiles on the same biological samples GSM21236 (in GDS817) versus GSM21242 (in GDS820); (b) Scatterplot of standardized profiles GSM21236/GSM21240 (in GDS817) versus GSM21242/GSM21246 (in GDS820); (c) Scatterplot of original expression profiles on different biological samples GSM31102 (in GDS855) versus GSM31127 (in GDS854); (d) Scatterplot of standardized profiles GSM31102/GSM31092 (in GDS855) versus GSM31127/GSM31117 (in GDS854). Joint work with X.J. Zhou (USC), J.C. Liu (USC)
Standardized profiles are consistent with the phenotype annotations Red Curve:different labs, same type of biological samples, same platform(Pearson correlations between GDS1372 and GDS1665) Blue curve: different labs, different biological samples, same platform(Pearson correlations between GDS1665 and GDS1917) Before Standardization After Standardization Joint work with X.J. Zhou (USC), J.C. Liu (USC)
Method highlights • Data Preparation • Standardizing the expression data to remove cross-lab and cross-platform incompatibilities (challenge 1). • Phenotypically annotating the collected human microarray experiments by Unified Language Medical System (UMLS) concepts (challenge 2). • UMLS also provides the language processing tool MetaMap to enable the automated mapping of text onto UMLS concepts • Processing the metadata has been one of the major efforts in recent Translational Bioinformatics research • Bayesian disease inference (challenge 3) • Bayesian belief network for refining the results (challenge 3) Joint work with X.J. Zhou (USC), J.C. Liu (USC)
Unified Medical Language System (UMLS) • UMLS consists of three major components: • Metathesaurus: > 1 million biomedical concepts from over 100 data sources. • Semantic Network: defines relationships between concepts. • Lexical resources: natural language processing tool. It can process text into the concepts.
UMLS Metathesaurus • Cluster synonymous terms into a single UMLS concept • Choose the preferred term • Assign the unique identifier Addison's disease SNOMED CT 363732003 Addison's Disease MedlinePlus T1233 Addison Disease MeSH D000224 Bronzed Disease SNOMED Intl 1998 DB-70620 Deficiency; corticorenal, primary ICPC2-ICD10 MTHU021575 Primary Adrenal Insufficiency MeSH D000224 Primary hypoadrenalism syndrome, Addison MedDRA 10036696 C0001403 Addison's disease
Dataset UMLS annotation construction http://www.ncbi.nlm.nih.gov/projects/geo/gds/gds_browse.cgi?gds=563 1. Take summary in dataset 2. Take PMID 3. Download MeSH headings from PubMed by PMID as follows: MH - Muscle Proteins/*genetics MH - Muscle, Skeletal/cytology/*pathology/physiology/physiopathology MH - Muscular Dystrophy, Duchenne/*genetics/*pathology MH - Oligonucleotide Array Sequence Analysis … etc. • 4. Parse both summary and MeSH headings by MetaMap to UMLS concepts as follows: • C0013264 Muscular Dystrophy, Duchenne • C0752352 Muscular Disorders, Atrophic • C0242692 Skeletal muscle structure • C0027868 Neuromuscular Diseases • … etc.
(1) Nervous system disorder (2) Neuromuscular Diseases (3) Myopathy (4) Musculoskeletal Diseases (5) Congenital, Hereditary, and Neonatal Diseases and Abnormalities (6) Genetic Diseases, Inborn (7) Genetic Diseases, X-Linked (8) Muscular Disorders, Atrophic (9) Muscular Dystrophies (10) Muscular Dystrophy, Duchenne Table 1. the phenotype annotation set for the dataset NCBI GEO GDS563 Joint work with X.J. Zhou (USC), J.C. Liu (USC)
Processing the metadata • Aronson, A.R. (2001) Effective mapping of biomedical text to the UMLS Metathesaurus: the MetaMap program. Proc AMIA Symp: 17-21. • Has been one of the major efforts in recent Translational Bioinformatics research • Butte AJ, Kohane IS.(2006) Creation and implications of a phenome-genome network. Nat Biotechnol.2006 Jan;24(1):55-62. • Shah NH, Jonquet C, Chiang AP, Butte AJ, Chen R, Musen MA. (2009) Ontology-driven indexing of public datasets for translational bioinformatics. BMC Bioinformatics. 2009 Feb 5;10 Suppl 2:S1.
To integrate the large amount of data on various diseases to build a diagnosis database such that users can rapidly search the disease profiles for expression similarities and further for disease annotations to the query sample of interest. We consider our diseasediagnosis question as aclassificationproblem, where each UMLSconcept represents an individual class, andall of the classes are organized in a hierarchy. The outcomeof our analysis is to categorize a standardized query dataset intoseveral classes in the hierarchy. This type of general setting is a so-called hierarchical multilabel classification (HMC) in the machine learning field.
Building Bayesian classifier for each disease class • We aim to infer • Difficulties include: • The association strength between different microarrays to the same disease class can vary greatly; • The distribution of the similarity scores sx,i is non-standard. Database phenotype-group M
To compute , we need model
Log-linear regression • guided by the following properties: • When all the rest are the same, • Larger means of scores should give larger ratios; • Bigger variances should give larger ratios; • Less skewness and kurtosis should give smaller ratios. Joint work with X.J. Zhou (USC), J.C. Liu (USC)
Making the annotation predictions For a query profilex, we will diagnose it with UMLS concept Ukif In our study, we set λto be 4.5. Joint work with X.J. Zhou (USC), J.C. Liu (USC)
We implemented a popular exact inference method, variable elimination, to infer Refining the inferred annotations • The UMLS concepts A, B, C, D, …, I are organized in a directed acyclic graph, with each node corresponds to a concept. • Given a query profile, and for each of the concept, we use A^, B^, … to denote the obtained bayesian annotation. • We note that for some nodes, the first round prediction is missing. By associating (conditional) probabilities with the DAG, we formulate our problem as a Bayesian Belief Network (BBN). Joint work with X.J. Zhou (USC), J.C. Liu (USC)
(1) Nervous system disorder (2) Neuromuscular Diseases (3) Myopathy (4) Musculoskeletal Diseases (5) Congenital, Hereditary, and Neonatal Diseases and Abnormalities (6) Genetic Diseases, Inborn (7) Genetic Diseases, X-Linked (8) Muscular Disorders, Atrophic (9) Muscular Dystrophies (10) Muscular Dystrophy, Duchenne Table 1. the phenotype annotation set for the dataset NCBI GEO GDS563 Joint work with X.J. Zhou (USC), J.C. Liu (USC)
7 23 21 22 3 1 12 11 18 19 20 15 13 10 9 2 4 5 6 16 8 17 13 9 8 12 11 14 10 4 7 3 6 2 5 1 Case study GDS2251: Comparison of myeloid leukemia cells to normal monocytes. Chromosome abnormality and translocation are highly related to myeloid leukemia. Bayesian prediction: Precision 59.1% recall 92.9% 1. Leukocytes, Mononuclear 2. monocyte 3. Bone Marrow Cells 4. Dysmyelopoietic Syndromes 5. Myeloid Leukemia 6. Nonlymphocytic Leukemia, Acute 7. Leukemia, Myelocytic, Acute 8. Immunoproliferative Disorders 9. Lymphoproliferative Disorders 10. Lymphoblastic Leukemia 11. Myeloid Cells 12. Phagocytes 13. Leukemia 15. Stem cells 16. Hematopoietic stem cells 17. Bone Marrow 18. Myeloid Progenitor Cells 19. Chromosomal translocation 20. Chromosome abnormality 21. neutrophil 22. Myeloproliferative disease 23. granulocyte • Subset 1: normal • Subset 2: myeloid leukemia “Bone Marrow” is not in the original set of Bayesian annotations but “Bone Marrow Cells” is. Therefore, “Bone Marrow” should be a correct annotation. • UMLS annotation: • Leukocytes, Mononuclear • monocyte • Bone Marrow Cells • Dysmyelopoietic Syndromes • Myeloid Leukemia • Nonlymphocytic Leukemia, Acute • Leukemia, Myelocytic, Acute • Immunoproliferative Disorders • Lymphoproliferative Disorders • Lymphoblastic Leukemia • Myeloid Cells • Phagocytes • leukemia • Malignant Neoplasms Myeloproliferative disease may evolve into myeloid leukemia. …etc.
Results: Leave-One-out Cross Validation Our method achieved an overall accuracy of 95% (precision 82% and recall 20%) The problem is analogous to other biological hierarchical multilabel classification problem, such as gene function prediction, which has achieved the best performance in mouse model at the recall rate of 20% and precision rate 41% (L. Pena-Castillo et al., Genome Biol, 2008). Joint work with X.J. Zhou (USC), J.C. Liu (USC)
Reduce the number of datasets Further accumulation of datasets would increase the power of our method. Joint work with X.J. Zhou (USC), J.C. Liu (USC)
Discussions • Power of this type of approach will increase daily with the continuous and rapid accumulation of genomics data in the public repositories. • Our diagnosis system is also promising in its potential to reveal unexpected disease connections, and further to construct novel phenome networks. • Better tools for text-mining are needed! Joint work with X.J. Zhou (USC), J.C. Liu (USC)
Discussions • Power of this type of approach will increase daily with the continuous and rapid accumulation of genomics data in the public repositories. • Our diagnosis system is also promising in its potential to reveal unexpected disease connections, and further to construct novel phenome networks. • Better tools for text-mining are needed! Joint work with X.J. Zhou (USC), J.C. Liu (USC)
UMLS concepts: Bone Marrow Cells Dysmyelopoietic Syndromes Hematopoietic stem cells Immunoproliferative Disorders Leukemia, Myelocytic, Acute Lymphoproliferative Disorders Myeloid Leukemia Nonlymphocytic Leukemia, Acute Stem cells leukemia The authors’ writing style affects the UMLS annotations. Joint work with X.J. Zhou (USC), J.C. Liu (USC)
Ongoing Work I • To further improve the method prediction power (Ci-Ren Jiang) • (focusing on the second stage) Applying a different way for a more thorough collaborative error correction along the disease hierarchy • Previous Bayesian Belief Network model is time consuming and only allows one-way information exchange
To integrate the large amount of data on various diseases to build a diagnosis database such that users can rapidly search the disease profiles for expression similarities and further for disease annotations to the query sample of interest. We consider our diseasediagnosis question as aclassificationproblem, where each UMLSconcept represents an individual class, andall of the classes are organized in a hierarchy. The outcomeof our analysis is to categorize a standardized query dataset intoseveral classes in the hierarchy. This type of general setting is a so-called hierarchical multilabel classification (HMC) in the machine learning field.
Ongoing Project II • Focusing on particular disease diagnosis by collaborating with specialized medical doctors/researchers, e.g., comparing bipolar disorder vs schizophrenia (Wayne Lee; Dr. Fei Wang, MD, PhD, Psychiatry, Yale University) • To investigate the usefulness/effectiveness of high-throughput molecular information in distinguishing between bipolar disorder and schizophrenia • To compare existing clinical predictors with predictors from microarray data
Acknowledgements • Dr. X. Jasmine Zhou (Molecular and Computational Biology, USC) • Dr. Jim Chun-Chih Liu (Molecular and Computational Biology, USC) • Dr. Ming-Chih Kao (Stanford Hospital, PhD, MD) • Dr. Ci-Ren Jiang (UCB) • Wayne Lee (UCB) • Dr. Fei Wang (Yale U)