How many transcripts does it take to reconstruct the splice graph?
10 likes | 176 Views
Introduction

How many transcripts does it take to reconstruct the splice graph?
E N D
Presentation Transcript
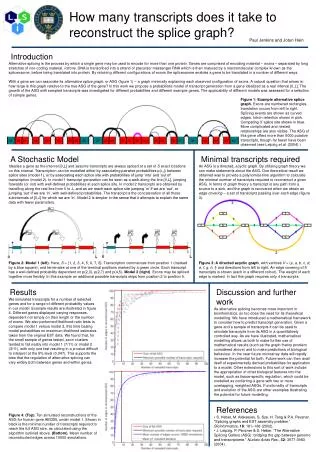
Introduction Alternative splicing is the process by which a single gene may be used to encode for more than one protein. Genes are comprised of encoding material – exons – separated by long stretches of non-coding material, introns. DNA is transcribed into a strand of precursor messenger RNA which is then matured by a macromolecular complex known as the spliceosome, before being translated into protein. By retaining different configurations of exons the spliceosome enables a gene to be translated in a number of different ways. With a gene we can associate its alternative splice graph, or ASG (figure 1) – a graph minimally explaining each observed configuration of exons. A natural question that arises is: how large is this graph relative to the true ASG of the gene? In this work we propose a probabilistic model of transcript generation from a gene idealized as a real interval [0,L]. The growth of the ASG with sampled transcripts was investigated for different probabilities and different example genes. The applicability of different models was assessed for a selection of sample genes. How many transcripts does it take to reconstruct the splice graph? Figure 1: Example alternative splice graph. Exons are numbered rectangles, translation occurs from left to right. Splicing events are shown as curved edges. Intron retention shown in pink. Competing 3’ splice site shown in blue. More complicated and nested relationships are also visible. The ASG of this gene offers more than 5000 putative transcripts, though far fewer have been observed (see Leipzig et al. (2004).) A Stochastic Model Idealise a gene as the interval [0,L] and assume transcripts are always spliced at a set of S exact locations on this interval. Transcription can be modelled either by associating pairwise probabilities p(i,,j) between splice sites (model 1), or by associating each splice site with probabilities of jump ‘into’ and ‘out of’ transcription (model 2). In model 1 transcript generation can be seen as a walk along the line [0,L], jumping forwards (or not) with well-defined probabilities at each splice site. In model 2 transcripts are obtained by travelling along the real line from 0 to L, and as we reach each splice site jumping ‘in’ if we are ‘out’, or jumping ‘out’ if we are ‘in’, with well-defined probabilities. The transcript is the concatenation of all those subintervals of [0,L] for which we are ‘in’. Model 2 is simpler in the sense that it attempts to explain the same data with fewer parameters. Minimal transcripts required An ASG is a directed, acyclic graph. By utilizing graph theory we can make statements about the ASG. One theoretical result we obtained was to provide a polynomial-time algorithm to calculate the minimal number of transcripts required to reconstruct a given ASG. In terms of graph theory a transcript is any path from a source to a sink, and the graph is recovered when we obtain an edge covering – a set of transcripts passing over each edge (figure 3). Paul Jenkins and Jotun Hein Figure 2: Model 1 (left). Here, S = {1, 2, 3, 4, 5, 6, 7, 8}. Transcription commences from position 1 (marked by a blue square), and terminates at one of the terminal positions marked by a green circle. Each transcript has a well-defined probability dependent on p(2,3), p(2,7) and p(4,5). Model 2 (right). Exons may be spliced together more flexibly. In this example an additional possible transcripts skips from position 2 to position 5. Figure 3: A directed acyclic graph, with vertices V = {s, a, b, c, d, e, f, g, h, t} and directions from left to right. An edge covering of 5 transcripts is shown (each in a different colour). The weight of each edge is marked. In fact this graph requires only 4 transcripts. Results We simulated transcripts for a number of selected genes and for a range of different probability values in our model. Example results are illustrated in figure 4. Different genes displayed varying responses, dependent not simply on their length or the number of exons. We also performed likelihood ratio tests to compare model 1 versus model 2, this time basing model probabilities on maximum likelihood estimates taken from the original EST data. We found that, for the small sample of genes tested, exon clusters tended to fall neatly into model 1 (7/11) or model 2 (3/11), with only one test resulting in a p-value difficult to interpret at the 5% level (0.047). This supports the idea that the regulation of alternative splicing can vary widely both between genes and within genes. Discussion and further work As alternative splicing becomes more important in bioinformatics, so too does the need for its theoretical modelling. We have introduced a mathematical framework to consider how to predict transcript generation. Given a gene and a sample of transcripts it can be used to simulate transcripts from its ASG in a quantitatively controlled way. As we have illustrated, mathematical modelling allows us both to make further use of mathematical results (such as the graph theory problem considered above) and to make predictions of biological behaviour. In the near future microarray data will rapidly increase the potential for both. Future work can then avail itself of experimentally derived probabilities for application to a model. Other extensions to this sort of work include the appropriation of other biological features into the model, such as tissue-specific regulation, which could be modelled as conferring a gene with two or more overlapping, weighted ASGs. Functionality of transcripts and evolution of the ASG are other examples illustrating the potential for future modelling. References • S. Heber, M. Alekseyev, S. Sze, H. Tang & P.A. Pevzner. “Splicing graphs and EST assembly problem.” Bioinformatics, 18: 181–188 (2002). • J. Leipzig, P. Pevzner & S. Heber. “The Alternative Splicing Gallery (ASG): bridging the gap between genome and transcriptome.” Nucleic Acids Res., 32: 3977-3983 (2004). Figure 4: (Top). Ten simulated reconstructions of the ASG for human gene ABCB5, under model 1. Shown in black is the minimal number of transcripts required to reach the full ASG size, as calculated using the algorithm outlined above. (Bottom). Mean number of reconstructed edges across 10000 simulations.





















