Download

1 / 67
960 likes | 2.09k Views
Inherited Kidney Diseases. Zehra Eren M.D. Nephrology Department. LEARNING OBJECTIVES. Recognize Renal cystic disorders - Autosomal dominant polycystic kidney disease - Autosomal recessive polycystic kidney disease - M edullary sponge kidney - Medullary c ystic kidney d isease

E N D
Inherited Kidney Diseases Zehra Eren M.D. Nephrology Department
LEARNING OBJECTIVES Recognize • Renalcysticdisorders -Autosomal dominant polycystickidneydisease -Autosomal recessive polycystic kidney disease -Medullaryspongekidney -Medullary cystickidney disease -Tuberoussclerosiscpmplex -vonHippel-Lindaudisease
LEARNING OBJECTIVES • Alport’sSyndrome • Thinbasementmembranedisease • Anderson-Fabrydisease • Nail-PatellaSyndrome • Sicle Cell Nephropathy
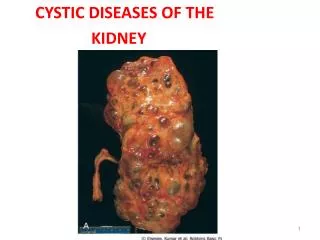
AUTOSOMAL DOMINANT POLYCYSTICKIDNEY DISEASE (ADPKD) • Epidemiology The most common renal hereditary disease,affects 1 in 400 to 1,000 live births Affects all races with equal frequence
Autosomal dominant polycystic kidney disease • Prevalence: 1:300 to 1:1000 • 90% of cases are inherited, 10% are sporadic • Only1 to 5% nephrons developed cysts • Cysts are in medulla and cortex • ADPKD causes symptoms in third or fourth decade • 50% of patients developing ESRD by age 60
AUTOSOMAL DOMINANT POLYCYSTICKIDNEY DISEASE (ADPKD) • Inheritance - Mutations in the polycystic kidney disease(PKD)1 gene:85%, located on the short armof chromosome 16 (16p.3.3), codes for a 4,304-amino-acid protein(polycystin 1) -Mutations in the PKD2 gene:15%, located on chromosome4 (4q.21.2), codes for a 968-amino-acid protein(polycystin 2)
ADPKD • Pathology - massive enlargement ofthe kidneys secondary to cyst growth anddevelopment • Diagnosis -Radiologic Radiological imaging is required(ultrasound is the current imaging modality ofchoice in at-risk individuals (positive familyhistory in a parent). -Genetic
ADPKD • Pathology - massive enlargement ofthe kidneys secondary to cyst growth anddevelopment • Diagnosis -Radiologic Radiological imaging is required(ultrasound is the current imaging modality ofchoice in at-risk individuals (positive familyhistory in a parent). -Genetic
Clinical Manifestations • Renal Chronicflankpain Acutepainindicates: infection (pyelonephritis- pyocyst) urinarytractobstruction suddenhemorrhageintocysts Hematuria Impairedrenalconcentratingability Nephrolitiasisin 15%to 20% Hypertensionin 75% adults
Clinical Manifestations • Extrarenal Hepatic cysts: Livercystspresentapproximately 10 yearsafterrenalcysts Intracranial aneurysms:Occur in 4% to 8% of symptomaticADPKD patients Cardiac disease:Mitral and aortic prolapse and regurgitation Diverticular disease: Hernias:abdominaland inguinal hernias (up to 45% ofADPKD patients)
AUTOSOMAL RECESSIVE POLYCYSTICKIDNEY DISEASE (ARPKD) • Epidemiology -Affects 1/10,000 to 1/40,000 individuals • Inheritance -Autosomal recessive disorder -Mutations in a single gene on the short armof chromosome 6p 21.1 • Pathology -The protein encoded by the PKHD1 gene iscalled polyductin or fibrocystin.
Diagnosis and Clinical Manifestations ● Prenatally: Oligohydramnios, enlarged kidneys,and lung hypoplasia with resultantPotter facies ● Infancy: Pneumomediastinum, pneumothorax,HTN, cardiac hypertrophy,endomyocardiofibroticcongestive heart failure, andrenal failure ● Older children: Hepatic fibrosis, portal HTN,and complications of variceal bleeding,thrombocytopenia, and anemia predominate
TUBEROUS SCLEROSIS COMPLEX (TSC) • Genetics -autosomal dominant pattern -high rate of spontaneousmutations (65% to 75% of patients) • TSC1 →9 (9q34), its protein product is calledHamartin • TSC2→16 (16p13.3),its protein product isnamedTuberin
MEDULLARY CYSTIC KIDNEY DISEASE(MCKD) • Rare inherited cystic disease, autosomal dominant inheritance: - MCKD1wasmappedtochromosome 1 (1q21) andaccountsfortheminority of cases - MCKD2wasmappedmorerecentlytochromosome 16 (16p12) andaccountsformutations in mostcases
MEDULLARY CYSTIC KIDNEY DISEASE(MCKD) • Pathology -normal- to smallsizedkidneys -cysts located atthe corticomedullaryjunction and in themedulla. However, the presence of cystsis not universal -Microscopically: diffuse tubulointerstitialinflammation, hypertrophiedand dilated tubules. Glomeruli areusually normal
Diagnosis and Clinical Course • Clinical features with a family history • The presence ofcysts supports the diagnosis but is not essential • Computed tomography scan is the mostsensitive technique for cyst detection • Polyuriaand polydipsia • Progressive renal failureultimately leads to ESRD
vonHippel-Lindaudisease • autosomal dominant syndrome manifested by a variety of benign and malignant tumors • VHL gene abnormality is present in about 1 in 36,000 newborns
von Hippel-Lindau disease • Clear cell RCCs occur in approximately 70 percent of VHL patients who survive to 60 years of age. • Annual imaging of the kidneys with MRI or CT is indicated to establish the diagnosis • For patients in whom an RCC is diagnosed, we recommend a nephron-sparing approach to remove lesions that are 3 cm or larger whenever possible
ALPORT’S SYNDROME OR HEREDITARYNEPHRITIS • Genetics -Prevalence of genetic mutation estimated at1 in 5,000 to 1 in 10,000. -Accounts for 1% to 2% of ESRD cases. -X-linked inheritance in almost all cases(85%) -Of the non–X-linked cases, most are autosomalrecessive
Alportsyndrome • Pathogenesis
ALPORT SYNDROME • Renal Manifestations -Hematuria: the characteristic clinical featureofAlport’s syndrome -Proteinuria • Extrarenal Manifestations -Sensorineural hearing loss -Ocular defects: anterior lenticonus -Leiomyomatosis of the esophagus and genitalia
THIN BASEMENT MEMBRANE DISEASE(TBMD) • Inherited renal disease of the GBM clinicallycharacterized by persistent microscopichematuria • inherited inan autosomal dominant fashion, is not accompaniedby extrarenal manifestations, andhas a benign course • The diagnosis is established by renal biopsy and electronmicroscopy