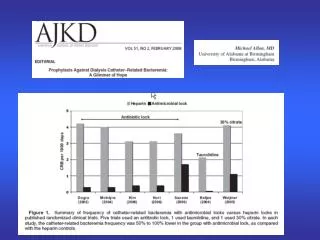
Download

1 / 30
330 likes | 648 Views
A Review for OCRA US RAC Study Group September 2005 Ginger Clasby, MS Promedica International gclasby@promedica-intl.com 714-799-1617 x 25. Investigational Device Exemptions 21 CFR Part 812. IDE About the Regulation.

E N D
A Review for OCRA US RAC Study Group September 2005 Ginger Clasby, MS Promedica International gclasby@promedica-intl.com 714-799-1617 x 25 Investigational Device Exemptions21 CFR Part 812
IDEAbout the Regulation • Provides procedures for conduct of clinical studies for unapproved devices • Exempts products from 510(k) or PMA approval requirements: • Registration, listing, interstate distribution • Labeling • GMPs • Applies to most studies to determine device safety & effectiveness
When IDE Application Not RequiredClinical Study Situations • Device legally marketed for indicated use • Marketing preference studies • Market seeding studies • Device is substantially equivalent to pre 1976 commercially distributed device • Device is non-significant risk & IRB has approved study • Many diagnostic devices • Veterinary devices • Custom device
Defining Device TypesSignificant or Non-Significant Risk? • Significant Risk: device has potential for serious risk to patient health, safety or welfare • Implants • Products supporting/sustaining life • Products with substantial role in diagnosing, curing mitigating or treating disease (or preventing impairment of human health) • Non-Significant Risk: all other devices
Defining Device TypesSignificant or Non-significant Risk? How to decide? Sponsor makes initial determination; IRB makes final call
Regulatory DifferencesSignificant vs. Non-significant Risk • Significant Risk Devices require FDA (IDE) & IRB approval prior to clinical study initiation • Non-Significant Risk Devices require only IRB approval prior to clinical study initiation • IDE presumed, unless FDA notified Sponsor that application is required
IDE Application ContentsAdministrative Details • Sponsor responsibility • 3 copies of submission required • All correspondence related to IDE sent by registered mail or by hand • IDE approval required prior to study initiation
IDE Application ContentsAn Overview • Sponsor name & address • Report of prior investigations & investigational plan • Description of methods, facilities, controls used for device manufacture, processing, packaging, storage & use • Names of proposed investigators & example of proposed investigator agreements • Certification that all investigators will sign agreement prior to study participation
IDE Application ContentsAn Overview • List of name, address & chairperson of each IRB reviewing study • If device will be “sold”, amount to be charged and rationale for why sale does not constitute commercialization • Environmental exemption/assessment • Proposed device labeling • “Caution – Investigational Device Limited by Federal (or US) Law to Investigational Use” • Copies of all informed consent documents
IDE ContentsReport of Prior Investigations • Reports of all prior clinical, animal & lab testing of device – should be adequate to justify proposed study • Bibliography of related publications • Summary of unpublished info • Non-clinical lab studies (in compliance with GLP)
IDE ContentsInvestigational Plan • Study purpose • Proposed protocol • Risk analysis • Device description • Monitoring procedures • Labeling • Consent materials
IDE ContentsInvestigational Plan • IRB info • Info on core labs, reading centers, etc. • Add’l records & reports
IDE Review/ApprovalFDA’s Considerations • Approval/disapproval decision • Rendered in writing • Rendered 30 days of FDA receipt • Approval may be denied/rescinded if: • Sponsor fails to comply with applicable regulations • Sponsor is non-responsive to requests for add’l info • Subject risks outweigh benefits • Unreasonable to proceed due to inadequacy of investigational plan, manufacturing or monitoring
Prohibited Practices Under IDESponsors & Investigators • Promotion of investigational product • Representing device as safe or effective for use in the study • Commercializing device by charging more than cost-recovery • Unduly prolonging study • Enrolling more subjects than necessary
IDE SupplementsNecessary When: • Changes made to investigational plan that may affect: • Scientific soundness of study • Rights, safety or welfare of study subjects • Changes in device design • Changes in device manufacturing • Addition of study investigators
“Treatment” IDEAppropriate When: • Device intended to treat or diagnose serious or life-threatening condition • No satisfactory alternative available • Controlled clinical trials in progress under IDE • Or when trials completed & FDA review of request to market is pending • Sponsor actively pursuing device marketing approval with FDA
Sponsor ResponsibilitiesSubpart C • Selection of qualified investigators & maintenance of ongoing communications • Appropriate monitoring of study • Special emphasis on device-related adverse events • Confirmation of IRB review/approval • Submission of IDE application to FDA • Prompt notification of significant new information to FDA, IRB
Sponsor ResponsibilitiesSubpart G – Records • Maintenance of records: • Study-related correspondence • Device shipment/disposition • Signed investigator agreements & financial disclosure info • Non-significant risk justification (if applicable) • Adverse device effects & complaints • Record retention ≥ 2 yr after study completion or date no longer required for PMA application • Record custody transferable with notice to FDA
Sponsor ResponsibilitiesSubpart G – Inspections • Must permit access by FDA employees to inspect facility where devices are held • Must permit access by FDA employees to inspect & copy study-related records
Sponsor ResponsibilitiesSubpart G – Reports • Unanticipated adverse device effects – 10 working days • Withdrawal of IRB approval – 5 working days • Withdrawal of FDA approval – 5 working days • Current investigator list – every 6 months • Progress reports – at least yearly • Device recalls & disposition – 30 working days
Sponsor ResponsibilitiesSubpart G – Reports • Study completion (significant risk) - 30 working days • Final report - 6 months • Use of device without written consent – 5 working days • Significant risk determination – 5 working days
IRB ResponsibilitiesSubpart D • Membership, duties & functions in accordance with 21 CFR 56 • Review of research • Initial review prior to study initiation • Significant risk determination, if necessary • Continuing review throughout study
IRB ResponsibilitiesSubpart G – Records & Reports • Records maintained in accordance with 21 CFR 56
IRB ResponsibilitiesSubpart G – Inspections • Must permit access by FDA employees to inspect & copy study-related records
Investigator ResponsibilitiesSubpart E • Compliance with: • Investigational plan • Investigator agreement(s) • Applicable FDA regulations • Protection of subject rights & welfare • Control of investigational devices • Obtaining consent in accordance with 21 CFR part 50 Repeated noncompliance with assigned responsibilities may result in investigator disqualification by FDA
Investigator ResponsibilitiesSubpart G – Records • Maintenance of records: • Study-related correspondence • Product receipt, use, or disposition • Subject case records • Protocol & amendments • Record retention ≥ 2 yr after study completion or date no longer required for PMA application • Record custody transferable with notice to Sponsor, FDA
Investigator ResponsibilitiesSubpart G – Inspections • Must permit access by FDA employees to inspect facility where devices are held • Must permit access by FDA employees to inspect & copy study-related records • Must permit access by FDA employees to inspect & copy records identifying subjects, if: • Suspicion that adequate consent was not obtained • Required investigator reports incomplete, inadequate, false
Investigator ResponsibilitiesSubpart G - Reports • Unanticipated adverse device effects – 10 working days • Withdrawal of IRB approval – 5 working days • Progress reports – at least yearly • Deviations from investigational plan – 5 working days • Device use without documentation of consent – 5 working days • Final report - 3 months
IDE RegulationReference Documents & Links (www.fda.gov/cdrh) • Guidance on IDE Policies & Procedures (1/1998) • Early Collaboration Meetings Under FDA Modernization Act (FDAMA), Final Guidance for Industry & CDRH Staff (2/2001) • Guidance on Significant & Non-Significant Risk Device Studies 7/25/86 (D86-1) • The Least Burdensome Provisions of FDA Modernization Act of 1997: Concept & Principles; Draft Guidance for FDA & Industry (5/3/2001)
IDE RegulationReference Documents & Links (www.fda.gov/cdrh) • FDA Guidance for Financial Disclosure by Clinical Investigators • FDA Guidance for IRBs & Clinical Investigators • FDA Guidance for Monitoring Clinical Investigations • FDA/ORA Compliance Program Guidance for Bioresearch Monitoring of Clinical Investigations • FDA CDRH Device Advice