
超重力在奈米粉體合成之特性 增加氣液沉澱反應之質傳 增加液液沉澱反應之混合
成核. 超重力反應結晶法合成奈 米粉體. 液體漿料. 進料 A 進料 B. nm 粒子. 長晶. Hi-G. Control induce time 2nm <100nm s~ms 200 分子 / 顆 奈米尺寸控制 7000~25000 分子 / 顆. 液體入口. 超重力在奈米粉體合成之特性 增加氣液沉澱反應之質傳
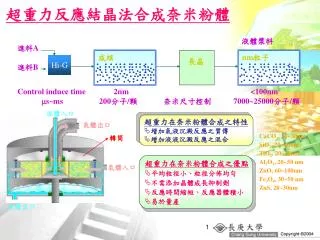

超重力在奈米粉體合成之特性 增加氣液沉澱反應之質傳 增加液液沉澱反應之混合
E N D
Presentation Transcript
成核 超重力反應結晶法合成奈米粉體 液體漿料 進料A 進料B nm粒子 長晶 Hi-G Control induce time 2nm <100nm s~ms 200分子/顆 奈米尺寸控制 7000~25000分子/顆 液體入口 超重力在奈米粉體合成之特性 • 增加氣液沉澱反應之質傳 • 增加液液沉澱反應之混合 氣體出口 CaCO3, 15~30nm SiO2, 20~40nm TiO2, 20~40nm Al2O3, 20~50 nm ZnO, 60~100nm Fe3O4, 30~50 nm ZnS, 20~30nm 轉筒 超重力在奈米粉體合成之優點 • 平均粒徑小、粒徑分佈均勻 • 不需添加晶體成長抑制劑 • 反應時間縮短、反應器體積小 • 易於量產 氣體入口 液體出口
奈米CaCO3合成 陳建峰,劉潤靜,沈志剛,宋繼瑞, 初廣文,”超重力反應沉澱法製備碳酸鈣的過程與形態控制“,過程工程學報,2(4),309-313,2002. Ca(OH)2懸浮液與CO2 氣體碳化反應時,其熱化學方程式可以表示為 根據水溶液的解離理論,在整個碳化反應過程中,存在以下四相:(1)氣相的CO2;(2)CO2溶解於Ca(OH)2懸浮液後形成的含有HCO3–、CO32–、H+、Ca2–、OH–等離子的混合溶液;(3)固相的CaCO3;(4)固相的Ca(OH)2。 其具體關係如下圖所示。從圖可知,碳化反應在氣–液–固多相體系中進行,它涉及到CO2氣體吸收、Ca(OH)2固體的溶解、CaCO3的沉澱及CaCO3粉體的成核、生長 和凝聚過程。 1. Stirred tank 2. Pump 3. Liquid distributor 4. RPB packing 5. Liquid outlet
奈米CaCO3合成 下圖為碳化過程的pH和導電度隨碳化時間的變化。從圖中可以看出,在通入二氧化碳氣體14 min時(約占整個碳化反應時間的80%),溶液的pH值只是在前4min時有所降低,此為碳化過程中的凝膠化階段。在此期間內,由於溶液過飽和度很高,CaCO3粉體成核過程中形成大量的線性中間體,構成網狀結構,從而使體系的黏度升高,並定向吸附在Ca(OH)2顆粒表面阻止Ca(OH)2的繼續溶解,使得溶液pH值下降,導電度也隨之下降,隨後很快恢復至初始值,基本保持不變,說明液相主體中Ca(OH)2濃度基本維持恆定,並接近其飽和濃度,此過程碳 化速率恒定,反應發生在氣、液界面的液膜中,過程速率受CO2的質傳控制。 在反應末期階段(約占整個碳化反應時間的20%),pH值迅速下降,溶液中Ca(OH)2濃度大大降低,溶解速率迅速減小,反應轉移至液、固界面的液膜中發生,過程速率受Ca(OH)2的溶解控制。在反應末期導電度略有回升,可能是有極少量的CaCO3與CO2和H2O發生反應生成Ca(HCO3)2從而使導電度略有回升。這和用普通的鼓泡 反應器製備CaCO3的過程特性一致。 另外,從圖中可以看出,由於在超重力環境下,CO2的質傳得到極大強化,大大縮短碳化反應時間(一般為10~20min),而普通碳化法制備CaCO3反應時間一般為60~75min,故採 用超重力碳化法可大大縮短碳化反應時間。 過程速率受Ca(OH)2的溶解控制
奈米CaCO3合成 在一定的氣液比、溫度和超重力水準、不同碳化時間測取pH值和導電度的同時,取樣製得粉 體樣品進行XRD分析,結果如右圖所示。圖中最上方的XRD圖譜為未碳化時的Ca(OH)2的懸 浮液經乾燥後粉體的XRD圖,與Ca(OH)2的標準譜圖是一致的,最下方的一條線是碳化結束時 漿液經乾燥後粉體的XRD圖譜,經對比與方解石型的標準譜圖一致,隨著碳化反應的進行, Ca(OH)2不斷地被消耗,峰值減弱,而新相在碳化反應不久很快產生,這與導電度和pH值隨碳化 時間的變化趨勢基本一致,由於反應液的凝膠化過程,使得溶液中的Ca(OH)2不能正常地解離而 導致pH值和導電度降低,隨著反應的進一步進行,方解石型碳酸鈣的晶相開始出現,反應液中 固相為Ca(OH)2和CaCO3兩相組成,當碳化至pH 為7左右時,Ca(OH)2全部轉化為結晶度很好 的方解石型CaCO3,故反應後的懸浮液乾燥後 XRD分析只有方解石型碳酸鈣的晶相出現。
奈米CaCO3合成 粉體的形貌是決定其應用性能的一個重要參數,應用體系不同,要求CaCO3的形狀不同,CaCO3晶體的形成過程包括兩個階段,即晶核的形成(nucleation)和晶核的長大(growth),已有不少文獻報導在攪拌槽中碳化反應的成核和生長速率,目前較為公 認的運算式為 Jn為成核速率[個/(m3.s)] G 為碳酸鈣晶體線性生長速率(m/s) Kn為成核速率常數 Kq為生長速率常數 其中n=4.2;Kn=107;q=2.0;Kq=8.0610-9 由此可見,碳酸鈣成核和生長都是非線性極強的快速過程,過飽和度微小的變化,將使成核和生長速率呈較大的變化,而成核速率變化更大。在超重力環境下,可極大地強化相間質傳和微觀混合,從而可以大大提高Ca(OH)2的溶解和CO2吸收速率,使反應體系中的[Ca2+]和[CO32–]瞬間得以迅速提高,過飽和度提高,導致體系CaCO3晶核數目大量 增加,製備出粒徑小且分佈均勻的立方形奈米CaCO3產品。
奈米CaCO3合成 圖(a)為在不添加任何晶形控制劑的條件下,採用超重力技術合成的奈米CaCO3粉體,平均粒徑為30nm, 且粒度分佈窄。 對於其他形貌的CaCO3粉體主要採用添加晶形控制劑來實現對CaCO3的形貌控制,其原理為CaCO3晶核的形成過程也是固體表面積激增的過程,從熱力學角度考慮,體系為了降低能量,即使形成CaCO3超微粉體也將凝聚成大的CaCO3粉體,若在碳化的某個階段加入晶形控制劑(或添加劑,通常是在凝膠化過程結束前加入),將發生兩個方面作用:一方面,由於形成的CaCO3微晶表面能很大、活性很高,可將晶形控制劑吸附於其表面,降低CaCO3微晶的表面自由能,使其能夠穩定存在,從而得到奈米CaCO3粉體;另一方面,可以調節CaCO3晶體的晶面能,控制各晶面的生長速率,某些晶面的生長受到抑制,使其他晶面沿擇優方向生長,從而得到不同形狀的CaCO3產品,右圖(b)~(h)為採用超重力法合成的其他 各種形狀的碳酸鈣TEM照片。
奈米CaCO3合成 ~對超重力碳化過程的研究表明,碳化反應前期為CO2吸收控制, 後期為Ca(OH)2溶解控制,反應前期有一明顯的凝膠化過程。 ~超重力法碳化時間較傳統碳化法碳化時間縮短4 倍以上。 ~在不添加任何晶體生長抑制劑的情況下,可以製備出平均粒徑為 15~30nm、分佈很窄、粒徑可控性好的奈米碳酸鈣。 ~在綜合控制一定操作參數和加入晶形控制劑的情況下能夠合成出 立方形、鏈鎖形、 紡錘形、薄片狀、玫瑰花瓣形、針狀、球形和 纖維狀等8種具有不同形態、不同粒徑的碳酸鈣粉體。
微米針狀CaCO3合成 朱萬誠,王玉紅,陳建峰,“超重力反應結晶法合成微細針狀碳酸鈣研究”,高校化學工程學報,5(16),560-564,2002 向混合溶液中加入Mg2+有利於文石型的形成,而不是穩定的方解石型相。具有文石型結晶相的碳酸鈣容易形成針狀外形。
微米針狀CaCO3合成 控制Ca(OH)2初始濃度在6.5%(約65gL-1)左右,總投料量3.6L。懸浮液pH值隨碳化時間變化曲線如右圖所示。對於不同濃度的添加劑(其他反應條件相同),pH值隨碳化時間變化顯示相同的趨勢。從圖可以看出,溶液的pH值在碳化的初始階段保持相對穩定,一定時間後出現突變,其後迅速下降;當停止通入CO2氣體後,pH值下降到一個最低點後有一定程度的反彈。在反應初期,Ca(OH)2懸浮液是過飽和溶液,有多少Ca(OH)2被反應掉就有多少Ca(OH)2固體粉體溶解到溶液之中,溶液中的OH–離子濃度保持恒定,因而這一時期pH值保持相對穩定;當固體Ca(OH)2粉體幾乎全部溶解後,反應掉的Ca(OH)2得不到補充,pH值開始突變,隨後急劇下降;停氣後,由於氣路中殘餘CO2繼續反應,pH值會再下降一些。由於反應過程中部分生成的CaCO3固體粉體包覆在Ca(OH)2粉體表面,隨著攪拌的持續進行,被包覆的Ca(OH)2粉體逐漸溶解,導致溶液pH值出現反彈現象。由圖可知,合成3mol微細針狀CaCO3約需碳化時間1h。與文獻報導的其他方法相比,合成等量(如3mol)針狀CaCO3,採用超重力反應結晶法所需碳化時間大大縮短,約為 文獻值的1/18。 其他反應條件相同時,隨著Ca(OH)2初始濃度的增加,反應時間將延長。Ca(OH)2初始濃度過大,將使得碳化初期凝膠化現象加劇,不利於反應的進行。實驗表明Ca(OH)2初始濃度對產品形貌沒有明顯的影響,Ca(OH)2初始濃度較適宜的範 圍是質量分率6.0%~7.5%。
微米針狀CaCO3合成 其他反應條件不變時,氣量增大,反應時間縮短,晶體成核速率加快,粒徑減小。實驗結果表明,在一定範圍內,氣量減小有利於針狀碳酸鈣的生成。氣液比對產品長徑比及形貌的影響如右圖所示。氣液比較適宜的範 圍是0.08~0.2。 旋轉填充床利用轉筒高速旋轉時產生的離心力,將Ca(OH)2懸濁液擊碎成為無數細小液滴。由於轉筒內液相高度分散、劇烈湍動,使氣液質傳相界面迅速更新,從而極大地強化氣液間的質傳。轉速增加,反應時間大大縮短,同時產品粒徑也將減小。轉速對產品長徑比及形貌的影響如右圖所示,可見轉速並非越大越好,較適宜的範 圍為900~1200 rpm。
微米針狀CaCO3合成 溫度對反應時間的影響不明顯,但對產品的粒徑影響較為明顯。溫度升高,Ca(OH)2溶解度降低,使得CaCO3生成速率下降,導致CaCO3過飽和度下降,成核速率降低,晶核數減少。因而,其他條件相同時,溫度升高,產品顆粒粒徑增大。實驗研究表明,溫度對CaCO3的晶形也產生較為明顯的影響。當溫度在13~30C之間時,CaCO3晶體多呈現立方形和鏈狀,有時還伴有不規則形態;當溫度在30~50C之間時,紡錘形出現的頻率較高,有時也會出現少量針形或類針形的晶體;當溫度升至55~75C之間時,會出現含量較高的針形。當溫度達到80~85C時,針形出現的機率減小,此時有較多的棒狀及少量紡錘形出現。溫度對產品長徑比及形貌的影響如右圖所示。綜合考慮,合成針狀碳酸鈣較適宜的溫 度範圍是55~75C。
微米針狀CaCO3合成 實驗表明,添加劑的加入對反應時間及產品粒徑均沒有明顯的影響。但MgCl2作為晶形控制劑,對合成針狀碳酸鈣產生決定性的作用。無添加劑時,產品中不會出現針狀晶體。當MgCl2濃度低於1.5%時,產品中幾乎也不會有針狀晶體出現。當高於10%時,產品中針形的含量一般很少。在一定範圍內,隨著添加劑MgCl2濃度的升高,針狀晶形減少,紡錘形增多;當其濃度很大,如高於20%時,幾乎不會出現針形或類針形的CaCO3晶體。添加劑濃度對產品長徑比及形貌的影響如右圖所示。晶形控制劑MgCl2較 適宜的濃度範圍是2.0%~6.0%。
微米針狀CaCO3合成 控制上述各操作變數分別在其較適宜的範圍內,採用旋轉填充床反應器合成的微細針狀碳 酸鈣TEM照片如右圖所示。 短軸及長徑比分佈如右圖所示。由圖可以看出,採用超重力反應結晶法所製備的微細針狀碳酸鈣具有較窄的短軸及長徑比分佈。在全部粉體中,短軸88%介於80~200nm之間,長徑比 82%介於12~20之間。
微米針狀CaCO3合成 ~用超重力反應結晶法,在旋轉填充床反應器中成功地合成平均 短軸為80~200nm、長徑比為12~20且短軸及長徑比分佈較窄的 微細針狀碳酸鈣。 ~只有在各影響因素較適宜的範圍內才能夠有效地合成微細針狀 碳酸鈣,即Ca(OH)2初始濃度為6.0%~7.5%,氣液比為 0.08~0.2,旋轉床轉速為900~1200rpm,反應溫度為55~75C, MgCl2濃度為2.0%~6.0%。 ~採用旋轉填充床反應器能夠極大地強化質傳和混合效果,大大 縮短反應時間,合成等量針狀CaCO3所需碳化時間約為文獻 值的1/18。
奈米BaTiO3合成 劉方濤,沈志剛,陳建峰,”超重力反應沉澱法(HGRP)製備納米BaTiO3的研究”,北京化工大學學報,30(1),5-8,2003 鋇鈦混合液中Ba/ Ti 莫耳比為1.07且[Ba2+]+ [Ti4+]=1.0mol/L;沉澱劑NaOH溶液濃度為6mol/L;鋇鈦混合液及NaOH溶液的流量均為90L/h;旋轉填充床轉速為1140rpm;反應溫度 為80C。 反應過程如左上圖所示,兩種反應物溶液分別裝入兩個儲槽中,經循環水預熱到一定溫度後,由兩個離心泵經質量流量計打入到旋轉填充床中,此時可將部分原料液循環回原料儲槽來輔助調節流量。原料液通過兩個開孔的噴嘴(見左下圖)噴到填充物上,液體在離心力的作用下沿填充物孔隙由轉筒內緣向轉筒外緣流動。在此期間原料液在填充物層中相互混合反應,在填充物外緣處甩到外殼上, 最後在重力作用下彙集到液體出口流出。
奈米BaTiO3合成 超重力反應沉澱法製得的粉體樣品採用乙醇作為分散劑,超音波分散30min,而後製成電子顯微鏡樣品,用電子顯微鏡透射成像得到TEM照片(圖a、b)。在相同的反應條件下,採用攪拌槽反應器製得鈦酸鋇粉體亦作透射電鏡分析作為對比(圖c)。從圖中可以看到,超重力法製得粉體粒徑為30~70nm且粒徑分佈較窄,鈦酸鋇粉體形態為接近單分散的球形。與攪拌槽中製備鈦酸鋇相比,超重力法合成的鈦酸鋇粉體粒徑明顯更小,且粉體形貌及分散性更好。這是因為超重力能極大地強化相間質傳,快速而均勻的實現反應物料的微觀混合,而且轉筒的高速旋轉所形成的高剪切應力削弱BaTiO3粉體間的團聚。超重力法樣品經BET法測試得到比表面積SBET為30.86m2/g,由dBET=6/×SBET (其中為BaTiO3 的理論密度,6.05g/cm3)計算得到BaTiO3粒徑為32.1nm,這與TEM分析結果基本吻合。
奈米BaTiO3合成 對超重力法製備粉體樣品進行X射線繞射分析,BaTiO3粉體的XRD圖譜如右圖。對照JCPDS標準資料,發現該法製得的是較純淨的立方相鈦酸鋇晶體 (JCPDS No.05-0626) 。 採用化學分析法對粉體的Ba/Ti比進行分析,分析結果見右表。分析結果表明鋇鈦混合液的Ba/Ti比預設為1.07時,製備粉體的Ba/Ti比為(1.000±0.005),達到2000年4月1日實施的化工行業標準(電子工業用高純鈦酸鋇)(HG/T 3587-1999)中規定的一等品鈦酸鋇粉體的 Ba/Ti 莫耳比要求(1.000±0.005) 。
奈米BaTiO3合成 採用AES-ICP對粉體樣品進行成分分析,結果見右表。從表中可以看到,除鐵含量偏高外,鈦酸鋇粉體中的幾種主要雜質元素的含量均低於行業標準中規定的雜質含量,且雜質含量指標明顯優於國外同類方法製備的鈦酸鋇粉體。說明採用超重力反應沉澱法可以製備優質高純的鈦酸鋇粉體。鐵含量偏高可能是由於反應裝置部分管路為不銹鋼管造成的,現正採用鈦管替換不銹鋼管,以改善這一狀況。鈉是反應的主要副產物,其含量可以通過用高 純水多次洗滌有效地降低。
奈米BaTiO3合成 ~將直接沉澱法和旋轉填充床結合起來的超重力反應沉澱法 是液相反應製備奈米粒體的一種有效方法。採用該法可以製 備出粒徑為30~70nm,粒徑分佈窄,分散性良好的奈米鈦酸 鋇粉體。 ~超重力反應沉澱法製備鈦酸鋇粉體的晶相為立方相,化學分 析得到Ba/Ti為(1.000±0.005),經AES-ICP進行雜質含量分 析,證明製得粉體為純度較高的鈦酸鋇粉體。 ~超重力反應沉澱法流程簡單,易於控制且沉澱物不需煅燒, 是一種極具工業應用潛力的製備方法。
奈米Al(OH)3合成 張鵬遠, 公延明, 陳建峰,”超重力碳分製備納米氫氧化鋁”,華北工學院學報,23(4),235-239,2002 稱取一定量的工業氫氧化鈉溶於水中,待完全溶解後,再稱取適量工業氫氧化鋁加入氫氧化鈉溶液中,保持二者的莫耳比(氫氧化鈉/氫氧化鋁)為1.0~1.5之間,加熱至氫氧化鋁完全溶解,得到粗鋁酸鈉溶液,用0.154mm的篩子過濾得濾液,即 得到一定濃度的過飽和SA溶液備用。 將製得的過飽和SA溶液加入旋轉填充床下部儲槽中,開離心泵循環,溶液流量經調節閥調節,由液體浮子流量計計量,並記錄初始溫度和pH值,待pH值相對穩定後,通入CO2氣體開始反應。保持整個溶液的溫度在30~60C之間(由於過程中放熱,若溫度超出給定範圍,可開冷卻水冷卻。)記錄溫度和pH值,當pH值到達11.5時,停止通 CO2氣,反應終止,產物由出料閥卸出。 產品料漿經真空過濾、洗滌,當洗滌液pH值到7時,停止洗滌,得濾餅,在乾燥箱中100~140C下乾燥10h,即得粉體產品。初次濾液由於含有大量鹼,可回收作為下次配料的原料。洗滌液可 用於清洗旋轉填充床。
碳酸化分解的實質是把二氧化碳通入鋁酸鈉溶液中,二氧化碳與溶液中的苛性鹼發生中和反應碳酸化分解的實質是把二氧化碳通入鋁酸鈉溶液中,二氧化碳與溶液中的苛性鹼發生中和反應 因此反應過程中鋁酸鈉溶液的穩定性隨著溶液苛性比值的急劇降低而被破壞,溶液隨即發生如下自發分解,析出氫氧化鋁沉澱 自發分解生成的NaOH 被繼續通入的CO2所中和,所以以上自發分解反應也得以繼續下去,直至所要求的分解程度, 但在碳酸化終結時, 通入的CO 2可與鋁酸鈉直接相互作用,而發生部分分解即 為了闡述碳酸化分解過程,可以將碳酸化分解成4個階段來說明,在第一階段內,溶液中的Al(OH)3含量不變, 整個過程是二氧化碳中和苛性鈉。因此溶液的苛性比下降,使溶液失去穩定性並開始分解;在第二階段中, 由於溶液具有最低的苛性比,因而Al(OH)3的過飽和度亦大,所以溶液自發分解的速度很快,但由於自發分解析出Al(OH)3的速度大於CO2中和苛性鈉的速度,此階段中溶液的苛性比值不斷地上升,直至此階段結束為止,苛性比可高達使溶液重新穩定的程度,使分解暫時停止;在第三階段內溶液的苛性比又開始下降直至溶液又失去其穩定性而進入第4 階段,以後的過程依此類推。當CO2氣體連續不斷均勻地通入鋁酸鈉溶液時,溶液中苛性鈉的含量隨之均勻地下降,而Al(OH)3的含量改變是不均勻的,是跳躍式的變化,苛性比的變化也是如此。這由圖中不同溫度下反應過程中pH值隨時間的變化曲線可以得 到很好的驗證,而這也與通常碳分分解過程相吻合。
奈米Al(OH)3合成 由右上圖為粉體在兩個不同的倍率中的TEM照片,由圖中可 知,該粉體的形態為薄片粒狀,其表觀粒徑小於50nm。 X射線繞射是鑒定物相最有效的方法之一,每種晶體都有一組特徵的晶面距值,其X射線繞射的分佈是一定的;每種晶體的原子排列也是一定的。因此,繞射線的相對強度也是一定的,即每一種晶體都有一套特徵繞射資料,並可把它作為定性鑑定物質所處相的依據,所以只要將XRD分析資料與標準值核對,就能準確知道物質的物相用X射線繞射儀作產品的X射線繞射分析,其X射線繞射 圖譜如右下圖所示。 將產品的X射線繞射圖與JCPDS標準卡片比較,可知其D 值在4.73174,4.36309,3.19397,2.22272處相應的繞射峰峰強分別為100.0,99.03,23.14,53.82;拜爾石(Bayerite sys)在D值為4.710,4.350,3.200,2.222相應的繞射峰峰強分別為90, 70,30,100,經比較可知二者大體相符,故可確定粉體晶 相為拜爾石,是三水氧化鋁的一種。
奈米Al(OH)3合成 由右圖可知,粉體初始失水温度在210C左右,200~600C之間共失水33.0%,比普通氫氧化鋁的初始失水温度略低,這是由於粉體相對較小,其 表面效應所引起的。 圖a為配製SA溶液時的濾渣的能譜圖,其主要含有Al、Na、Fe、Ca等金屬元素,圖b為最終碳分製備產品的能譜圖,由圖中可以看出,最終產品中未發現 有其他金屬離子,產品的純度相對較高。
奈米Al(OH)3合成 旋轉填充床的氣液質傳係數比傳統填充床高1~3個數量級,這非常有利於在氣液反應和沉澱反應中產生更高的過飽和濃度,而對於碳分分解過程來說,在旋轉填充床中,過飽和的SA溶液通過填充物後被分散成極細小的液滴、液絲和薄膜,這導致液體強烈微觀混合,爆發性析出大量的細小、均勻的氫氧化鋁晶核,且時間短,由結晶理論可知,可製備出超細氫氧化 鋁,因此,在理論上說旋轉填充床是製備奈米粉體的理想反應器。 通過在超重力場中碳分分解,成功製備出奈米氫氧化鋁,其表觀粒徑小 於50 nm ,實驗結果顯示: (1)超重力碳分法製備奈米氫氧化鋁的基本反應規律與通常碳分法一致; (2)經過進一步簡單放大研究表明,其流程放大簡單,放大效應小,流程 控制簡單,條件要求不苛刻,與傳統的製備方法相比較具有較好的應 用價值。
奈米SiO2合成 劉海弟,賈宏,郭奮,郭鍇,”超重力反應沉澱法製備白炭黑的研究”,無機鹽工業,35(1),13-15,2003 來自鋼瓶的二氧化碳和來自壓縮機的空氣經計量後進入旋轉填充床與事先配製好絮凝劑、分散劑的矽酸鈉水溶液反應。矽酸鈉水溶液由泵驅動在填充物中循環。熱水由水循環泵驅動在恒溫水浴和反應器夾套間循環,以保持反應溫度在預定範圍內。反應過程中定時取樣分析pH。當pH不再隨時間變化時,停止通氣並取反應產物置於燒瓶內,加酸調節pH到設定值,保溫老化。老化時間為設定值。老化後,抽濾、洗滌料漿產物,於100~105C下恒溫乾燥。最後 研磨、過篩(38m),製得二氧化矽粉體樣品。 矽酸鈉水解: Na2O·nSiO2+(2n+1)H2O 2NaOH+nSi(OH)4 碳酸鈉生成: 2NaOH+CO2 Na2CO3+H2O 矽酸脱水聚合: mSi(OH)4 m(SiO2·nH2O)+(2m-nm)H2O Si-OH+HO-Si Si-O-Si +H2O
T=85-90℃ V(纯CO2)=0.8m3/h R=1200rpm 奈米SiO2合成 右圖是在旋轉填充床中,以矽酸鈉和二氧化碳為原料,沉澱法製備白炭黑反應過程中反應液pH的變化情況。從圖可以看出,整個碳化反應分成明顯的3個階段:反應誘導階段、反應階段和反應結束階段。反應階段反應速度很快,其他兩個階段呈平緩趨勢。整 個反應只需20min。 左圖是矽酸鈉溶液濃度、絮凝劑加入量、分散劑種類及用量、老化時間、旋轉填充床轉速、反應溫度、CO2氣量一定的情況下,酸化、老化pH對產品粒徑的影響圖。從左圖的曲線變化趨勢來看,隨著老化pH的減小產品粉體粒徑有減小的趨勢,這可能是因為粉體電位變高,粉體間不易發生團聚,而且老化過程可以去除二氧化矽粉體團內部所吸附的水合碳酸根離子,從而將粉體團“切開”,達到細化粉體的作用。老化pH也影響最終產品的pH而該值是衡量產品是否符合國標的指標之一。比如在作為橡膠補強劑時,酸性的二氧化矽使混煉膠延遲硫化;鹼性二氧化矽則促進橡膠硫化,易使橡膠料發生焦燒。因此最好使二氧化矽中性,pH在7左右為宜。結果顯示老化pH為4~5時,最終產品pH在6.3~7.5。另外,二氧化矽酸性溶液比鹼性溶液穩定性好,較不易轉變成凝膠。一般來說,老化pH視 用戶對最終產品pH的要求而定,但不宜小於3。
T=85-90℃ V(纯CO2)=0.8m3/h R=1000rpm 奈米SiO2合成 從右圖可以看出,轉速的提高導致反應液pH變化曲線的斜率變大,這同時也說 明反應速率隨轉速的提高而增加。 當旋轉填充床轉速、絮凝劑用量、分散劑種類及用量、酸化、老化pH、老化時間、反應溫度、CO2氣量一定的情況下,矽酸鈉溶液初始濃度的變化對產品粒徑的影響見右圖。矽酸鈉溶液的初始濃度影響在原料液的初始密度上,濃度越高密度越大。原料初始濃度的降低有利於減小產品粒徑。這是因為初始濃度的提高並沒有使成核數目發生較大的變化,主要消耗於粉體的生長,因而粉體粒徑逐漸增大。矽酸鈉的黏度隨濃度的增加而升高,黏度過高不利於CO2的吸收操作,另外也容易發生凝膠,影響反應控制。結果顯示當矽酸鈉水溶液密度大於1075kg/m3後,凝膠化問題不易控制,可操作條件範圍窄。雖然低濃度下合成的產品粒子粒徑小且單分散性能較好,但反應物濃度過低導致生產能力低,單位時間內單位設備容量下的生產效率低。綜合考慮取矽酸鈉水溶 液初始密度為1051~1075kg/m3。
50nm 产品粒径(nm) 奈米SiO2合成 產品粒子多呈球形,平均粒徑小於30nm。 約95%的粒子在15~30nm之間,平均粒徑約為22nm。 超重力法製備二氧化矽的過程的反應機制和常規碳化法並無不同,但由於旋轉填充床的優點使該反應過程有不同於常規碳化法的特徵:(1)在旋轉填充床中,反應料液以液滴、液絲及液膜的形式高速通過填充物,其表面更新很快和逆流經過的氣體發生很好的氣液接觸,二氧化碳吸收充分;(2)因為高速旋轉填充物的切削和撞擊,填充物中存在大量的小液滴甚至細霧,在適當反應溫度下,液滴的汽化非常劇烈,這大大促進反應產生的矽酸的脫水,使二氧化矽粉體的長大加快,然而由於液滴微細,粉體無法長得太大。這就是超重 力法反應時間很短(20min左右)但所得產品仍然微細的原因。
奈米ZnO合成 毋偉,張新軍,陳建峰,王國全,”超重力法納米氧化鋅的製備表徵及其應用”,北京化工大學學報,32(2),25-28,2005 右圖為不同操作條件下實驗室製備的兩種奈米氧化鋅TEM照片,圖a為中間體未經老化直接過濾洗滌煅燒後得到的氧化鋅樣品的TEM照片,外觀為棒狀,寬約20nm,圖b是反應料液經12h老化後洗滌、過濾、煅燒得到的氧化鋅樣品的TEM照片,外觀近似為球形,其粒徑約為30nm。這說明奈米氧化鋅粉體形狀很大程度受中間體粉體形狀的影響,不經老化的中間體在空間的生長是不均衡的和不穩定的。在同一液體介質中,粉體越小,溶解度越大,在老化過程中,間體中小粉體逐漸溶解,達到過飽和度後在大粉體表面成核生長,得到均一穩定的中間體粉體形狀和大小,中間體的形狀和大小影響到最後得到的奈米氧化鋅的粉體形貌及大小。不同老化時間與奈米氧化鋅粉體形貌之間的關係還有待進一步研究。所得奈米氧化鋅分散性良好,不同形狀的奈米氧化鋅可以滿足不同的用 途。例如橡膠工業多用球狀,油漆多選用柱狀。 (a) (b)
奈米ZnO合成 紫外線是太陽光線中波長最短的一種,其能量約占太陽光線總能量的6%,作用於皮膚的紫外線分為短波紫外線(UVC:200~290nm)、中波紫外線(UVB:290~320nm)、長波紫外線(UVA:320~400nm)。對人體皮膚影響最大的UVB和UVA, 用不同粒徑的球形氧化鋅測其紫外-可見光曲線如右圖所示。 從右圖中可以看出,該法製得的奈米氧化鋅對250~400nm波長範圍的紫外線有很強的吸收作用,與奈米二氧化鈦相比不但吸收作用強,而且吸收範圍寬,對UVA和UVB均有吸 收作用。粒徑越小,吸收作用越強。 對於奈米粉體來說,低溫下電子能階是不連續的,符合 式中為能階間距;Ef為費米能階;N為總電子數;V為粉體的體積。可見粉體的體積越小,電子能階間隔越大,對小波長紫外線吸收能力越強。奈米氧化鋅的粒徑與紫外可見光吸 收性能的關係符合這一規律。
奈米ZnO合成 右圖為從旋轉填充床製備出且經充分洗滌乾燥後的中間體熱失質量-差熱分析曲線。假定其分解溫度為200C,則分解失質量率約為20%,與氫氧化鋅的化學計量失質量基本相當,可以推測在奈米氧化鋅 的製備過程中主要發生如下反應 將製得的球形氧化鋅進行XRD分析。由左圖可以看出,得到的奈米氧化鋅XRD峰尖銳,表明其結晶性好,晶形完整。並且與標準氧化鋅譜圖34-1451完全吻合,為銳鋅型(zincite syn)氧化鋅。由謝樂公式D=k/cos(式中k為Scherrer常數;為X射線特徵波長;為繞射線剖面的半寬度;為布拉格角)。計算得出粉體的晶粒徑D約為23nm,比從TEM照片 測得的數值稍小。
奈米ZnO合成 將超重力法製得的奈米氧化鋅和從它處得到的微米氧化鋅分別添加到PVC糊中,作PVC糊的發泡促進劑,下表是實驗所得結果。奈米氧化鋅對PVC糊發泡有明顯的促進 作用,發泡率明顯高於添加微米級氧化鋅和未添加任何氧化鋅的PVC糊樹脂。
奈米Mg(OH)2合成 宋雲華,陳建銘,劉立華,郭奮,”超重力技術製備納米氫氧化鎂阻燃劑的應用研究”,化工礦物與加工, 2004(5),19-23 本實驗採用液液法製備奈米氫氧化鎂阻燃劑。鹵水放入槽2中,氨水放入槽1中,鹵水進入旋轉填充床與一定質量分率的氨水在一定轉速下接觸並發生反應,生成鹼式氯化鎂沉澱,通過離心力的作用將其甩出後進入槽1,攪拌槽和旋轉填充床之間用泵循環,保溫熱處理一定時間後確保鹼式氯化鎂完全轉化為氫氧化鎂時結束反應。
奈米Mg(OH)2合成 由右圖的TEM照片可以看出,採用超重力技術製備的氫氧化鎂為70nm的六方型片狀物。研究分析顯示,在超重力環境下,不同大小分子間的分子擴散和相間質傳過程均比常重力場下的要快得多,氣液兩相在比地球重力場大數百倍至千倍的超重力環境下的多孔介質中產生流動接觸,巨大的剪切力將液體撕裂成奈米級的膜、絲和滴,產生巨大的和快速更新的相界面,使相間質傳速率比傳統的塔器中提高1-3個數量級,微觀混合和質傳過程得到極大強化。在反應過程中,料液通過液體分佈器噴向旋轉填充床轉筒內, 在離心力的作用下由旋轉填充床內緣流向外緣。 由XRD分析結果可以看出,由超重力技術製備的奈米氫氧化鎂符合Brucite氫氧化鎂標準位元峰資料,形狀為六方型,且特徵繞射峰強度高、 峰形尖銳,說明產物結構規整性強。
奈米Mg(OH)2合成 將奈米氫氧化鎂濾餅配成5%~7%的漿料,升溫至85C左右,乳化機的攪拌速度調至5000rpm,滴加2%~5%(以乾粉質量為基準)的改性劑,持續0.5h,過濾洗滌,於110C下 烘乾,過250目篩備用。 由右表可以看出,改性粉體的比表面積比未改性粉體有一定程度的增加。這是因為未改性奈米氫氧化鎂粉體的粒徑較小,表面能較高,極易發生團聚,從而降低它的比表面積;改性粉體雖然粒徑也較小,但由於其表面包覆一層有機物,從而降低表面能,防止團聚現象的發 生,奈米氫氧化鎂粉體的分散性大為改善。 對粉體潤濕性好的液體,粉體能在其中很好地分散,粒體不易黏結聚集,沉降速度慢;反之,粒體分散性差,易聚集,沉降速度快。由右圖可以看出改性後比改性前的粉體在液體石蠟中的沉降速度明顯變慢,這是由於表面改性使得奈米氫氧化鎂粉體表面包覆一層有機物,從而和有機介質-液體石蠟有很好的相容性,能在其中較好分散的緣故。從沉降速度來看,以 硬脂酸鋅改性效果最佳。 稱取5g改性前後的奈米氫氧化鎂粉體置於50mL的柱塞量筒內,加液體石蠟至50mL 刻度,靜置,讀取不同時間固體的體積
奈米Mg(OH)2合成 由右表資料可以看出,未改性粉體的吸油值較高,對DOP的無效吸收較嚴重,而改性後的奈米氫氧化鎂粉體的吸油值明顯下降。這說明表面改性使得其在軟質PVC體系中應用時對DOP增塑劑的無效吸收量減少,避免因此造成的加工過程中聚合物的分解,同時也可以降低成本。從吸油值角度考 慮,以硬脂酸鋅改性效果最佳。 將DOP滴加到改性前後的奈米氫氧化鎂粉末中,用調墨刀研壓使之成團不散, 測定吸油量。 由左表的實驗資料可以看出,改性體系比未改性體系和純基體樹脂的氧指數有一定程度的提高,奈米氫氧化鎂的表面改性處理確實改善體系的阻燃性能;機械力學性能比純基體樹脂有所下降,但比未改性體系要好。這是由於奈米氫氧化鎂的表面改性在一定程度上提高疏水性能,改善它在PVC體系中的分散性,因此提高體系的阻燃性能,同時降低對體系機械力學性能的影響。隨著所選用的表面改性劑的不同,改善程度也不盡相同。在本實驗條件下硬脂酸 鋅改性粉體的應用效果較好。
奈米Mg(OH)2合成 作為無機添加型阻燃劑,隨著奈米氫氧化鎂粉體用量的增加,體系氧指數逐漸增大,拉伸強度和斷裂伸長率逐漸降低。在PVC中添加100份的奈米氫氧化鎂粉體,其氧指數由純基體樹脂的25.5上升到30.1,達到很好的阻燃效果,但拉伸強度由純基體樹脂的23.588MPa下降到13.369MPa,下降幅度很大,難以達到實際使用要求。綜合考慮對阻燃性能和機械力學性能的影響,在本實驗條件下奈米氫氧化鎂阻燃劑 的最佳添加量為40份。
作業四 若用超重力反應結晶法合成奈米銀粉體,請列出一種可能使用的反應系統。