Download

1 / 55
560 likes | 717 Views
This presentation by C. Titus Brown, Assistant Professor at Michigan State University, delves into innovative methods for assembling large, complex metagenomes. Highlighting notable challenges in genome assembly, such as the issues of repeat-rich genomes and sequencing biases, Brown discusses case studies involving the H. contortus genome and lamprey transcriptome. The talk emphasizes the necessity of advanced filtering techniques, the importance of assembly in improving bioinformatics analysis, and the potential to unlock novel insights into microbial ecology through sophisticated sequencing approaches.

E N D
The pro-shotgun-assembly talk. C. Titus Brown Assistant Professor CSE, MMG, BEACON Michigan State University ctb@msu.edu
Collaborators Acknowledgements Lab members involved • Adina Howe (w/Tiedje) • Jason Pell • ArendHintze • RosangelaCanino-Koning • Qingpeng Zhang • Elijah Lowe • LikitPreeyanon • JiarongGuo • Tim Brom • KanchanPavangadkar • Eric McDonald • Jordan Fish • Chris Welcher • Jim Tiedje, MSU • Billie Swalla, UW • Janet Jansson, LBNL • Susannah Tringe, JGI Funding • USDA NIFA; NSF IOS; BEACON.
Open, online science All of the software and approaches I’m talking about today are available: Assembling large, complex metagenomes arxiv.org/abs/1212.2832 khmer software: github.com/ged-lab/khmer/ Blog: http://ivory.idyll.org/blog/ Twitter: @ctitusbrown
Note: I am phylogenetically unconstrained… • Chordate mRNAseq (Molgula + lamprey + chick) • Nematode genomics • Soil metagenomics …but so far not microbial euks, specifically.
My goals in this work • Interested in genes & genomes: function & evolution, but not as much taxonomy. • Little or no marker work (16s/18s) • Develop lightweight prefiltering techniques for other tools. • Software & methods => democritize data analysis.
I am unambiguously pro-assembly. • Short-read analysis can be misleading; need more work like Doc Pollard’s showing where/why! • Assembly reduces the data size, increases boinformatic signal, and eliminates random errors. • The general mental frameworks (OLC or DBG) underpin virtually all sequence analysis anyway, note. • So, why not? • Assembly is HARD, SLOW, TRICKY. • Assemblies may MISLEAD you. • Assembly is a STRINGENT FILTER on your data <=> heuristics.
There is quite a bit of life left to sequence & assemble. http://pacelab.colorado.edu/
Challenges of (micro-)euks • Genomes are large and repeat rich. • Diploidy and polymorphism will confuse assemblers. • Note: very problematic in tandem with repeats. • Nucleotide bias => sequencing bias. • Scarce samples => amplification techniques => sequencing bias. All of these confound assembly. Can we “fix”?
Three illustrative problem cases • H. contortus genome assembly. • Lamprey reference-free transcriptome assembly. • Soil metagenome assembly.
The H. contortus problem A sheep parasite. ~350 Mbp genome Sequenced DNA 6 individuals after whole genome amplification, estimated 10% heterozygosity(!?) Significant bacterial contamination. (w/Robin Gasser, Paul Sternberg, and Erich Schwarz)
H. contortus life cycle Refs.: Nikolaou and Gasser (2006), Int. J. Parasitol. 36, 859-868; Prichard and Geary (2008), Nature 452, 157-158.
The power of next-gen. sequencing: get 180x coverage ... and then watch your assemblies never finish Libraries built and sequenced: 300-nt inserts, 2x75 nt paired-end reads 500-nt inserts, 2x75 and 2x100 nt paired-end reads 2-kb, 5-kb, and 10-kb inserts, 2x49 nt paired-end reads Nothing would assemble at all until filtered for basic quality. Filtering let ≤500 nt-sized inserts to assemble in a mere week. But 2+ kb-sized inserts would not assemble even then. Erich Schwarz
So, problem 1: nematode H. contort Highly polymorphic Whole genome amplification Repeat ridden => Assemblers DIE HORRIBLY.
The lamprey problem. • Lamprey genome is draft quality; low contiguity, missing ~30%. • No closely related reference. • Full-length and exon-level gene predictions are 50-75% reliable, and rarely capture UTRs / isoforms. • De novo assembly, if we do it well, can identify • Novel genes • Novel exons • Fast evolving genes • Somatic recombination: how much are we missing, really?
Sea lamprey in the Great Lakes • Non-native • Parasite of medium to large fishes • Caused populations of host fishes to crash Li Lab / Y-W C-D
Lamprey transcrpitome Started with 5.1 billion reads from 50 different tissues. No assembler on the planet can handle this much data.
So, problem 2: lamprey mRNAseq Must go with reference-free approach. TOO MUCH DATA.
Soil metagenome assembly • Observation: 99% of microbes cannot easily be cultured in the lab. (“The great plate count anomaly”) • Many reasons why you can’t or don’t want to culture: • Syntrophic relationships • Niche-specificity or unknown physiology • Dormant microbes • Abundance within communities Single-cell sequencing & shotgun metagenomics are two common ways to investigate microbial communities.
Investigating soil microbial ecology • What ecosystem level functions are present, and how do microbes do them? • How does agricultural soil differ from native soil? • How does soil respond to climate perturbation? • Questions that are not easy to answer without shotgun sequencing: • What kind of strain-level heterogeneity is present in the population? • What does the phage and viral population look like? • What species are where?
Scaling challenges in metagenomics (and assembly, more generally) • It is difficult to even achieve an assembly for the volume of data we can easily get. (Also see: ARMO project, ~2 TB of data.) • Most current assemblers are quite heavyweight, perhaps partly because they are written by people with large resources. • This fails given scaling behavior of sequencing.
So, problem 3: soil metagenomics TOO MUCH DATA. BAD SCALING.
Approach: Digital normalization(a computational version of library normalization) Suppose you have a dilution factor of A (10) to B(1). To get 10x of B you need to get 100x of A! Overkill!! This 100x will consume disk space and, because of errors, memory. We can discard it for you…
Digital normalization approach A digital analog to cDNA library normalization, diginorm: Reference free. Is single pass: looks at each read only once; Does not “collect” the majority of errors; Keeps all low-coverage reads; Smooths out coverage of regions.
Coverage before digital normalization: (MD amplified)
Coverage after digital normalization: Normalizes coverage Discards redundancy Eliminates majority of errors Scales assembly dramatically. Assembly is 98% identical.
Wait, that works?? Note, digital normalization is freely available, with lots of tutorials. Derived approach now part of Trinity (Broad mRNAseq assembler). It is, ahem, still unpublished, but available on arXiv: arxiv.org/abs/1203.4802
1. H. contort after digital normalization • Diginorm readily enabled assembly of a 404 Mbp genome with N50 of 15.6 kb; • Post-processing with GapCloser and SOAPdenovo scaffolding led to final assembly of 453 Mbp with N50 of 34.2kb. • CEGMA estimates 73-94% complete genome. • Diginorm helped by: • Suppressing high polymorphism, esp in repeats; • Eliminating 95% of sequencing errors; • “Squashing” coverage variation from whole genome amplification and bacterial contamination
H. contort after digital normalization • Diginorm readily enabled assembly of a 404 Mbp genome with N50 of 15.6 kb; • Post-processing with GapCloser and SOAPdenovo scaffolding led to final assembly of 453 Mbp with N50 of 34.2kb. • CEGMA estimates 73-94% complete genome. • Diginorm helped by: • Suppressing high polymorphism, esp in repeats; • Eliminating 95% of sequencing errors; • “Squashing” coverage variation from whole genome amplification and bacterial contamination
Next steps with H. contortus Publish the genome paper Identification of antibiotic targets for treatment in agricultural settings (animal husbandry). Serving as “reference approach” for a wide variety of parasitic nematodes, many of which have similar genomic issues.
2. Lamprey transcriptome results Started with 5.1 billion reads from 50 different tissues. Digital normalization discarded 98.7% of them as redundant, leaving 87m (!) These assembled into more than 100,000 transcripts > 1kb Against known full-length, 98.7% agreement (accuracy); 99.7% included (contiguity)
Evaluating de novo lamprey transcriptome Estimate genome is ~70% complete (gene complement) Majority of genome-annotated gene sets recovered by mRNAseq assembly. Note: method to recover transcript families w/o genome… (Includes transcripts > 300 bp)
Next steps with lamprey • Far more complete transcriptome than the one predicted from the genome! • Enabling studies in – • Basal vertebrate phylogeny • Biliary atresia • Evolutionary origin of brown fat (previously thought to be mammalian only!) • Pheromonal response in adults
Additional Approach for Metagenomes: Data partitioning(a computational version of cell sorting) Split reads into “bins” belonging to different source species. Can do this based almost entirely on connectivity of sequences. “Divide and conquer” Memory-efficient implementation helps to scale assembly. Pell et al., 2012, PNAS
Partitioning separates reads by genome.Strain variants co-partition. When computationally spiking HMP mock data with one E. coli genome (left) or multiple E. coli strains (right), majority of partitions contain reads from only a single genome (blue) vs multi-genome partitions (green). * * Adina Howe Partitions containing spiked data indicated with a *
Assembly results for Iowa corn and prairie(2x ~300 Gbp soil metagenomes) Putting it in perspective: Total equivalent of ~1200 bacterial genomes Human genome ~3 billion bp Adina Howe
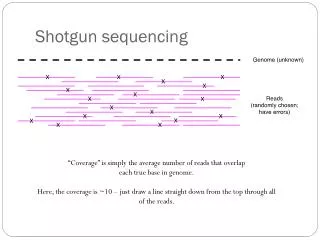
…but high coverage is needed. Low coverage is the dominant problem blocking assembly of your soil metagenome.
Strain variation? Can measure by read mapping. Of 5000 most abundant contigs, only 1 has a polymorphism rate > 5% Top two allele frequencies Position within contig
Overconfident predictions • We can assemble virtually anything but soil ;). • Genomes, transcriptomes, MDA, mixtures, etc. • Repeat resolution will be fundamentally limited by sequencing technology (insert size; sampling depth) • Strain variation confuses assembly, but does not prevent useful results. • Diginorm is systematic strategy to enable assembly. • Banfield has shown how to deconvolve strains at differential abundance. • Kostas K. results suggest that there will be a species gap sufficient to prevent contigmisassembly. • Even genes “chimeric” between strains are useful.