DIY DNA Extraction from Split Peas: Step-by-Step Guide
Learn how to extract DNA from split peas at home using simple ingredients and equipment. Follow step-by-step instructions for successful DNA extraction. Understand the principles behind the process and different techniques involved.

DIY DNA Extraction from Split Peas: Step-by-Step Guide
E N D
Presentation Transcript
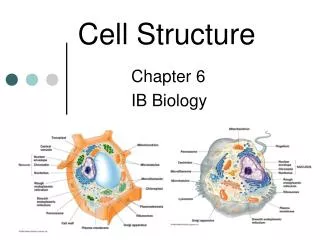
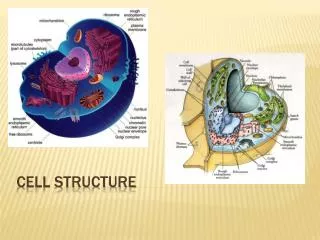
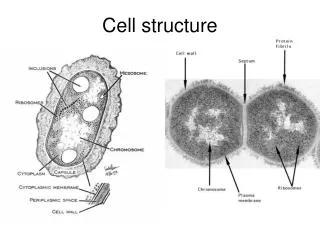
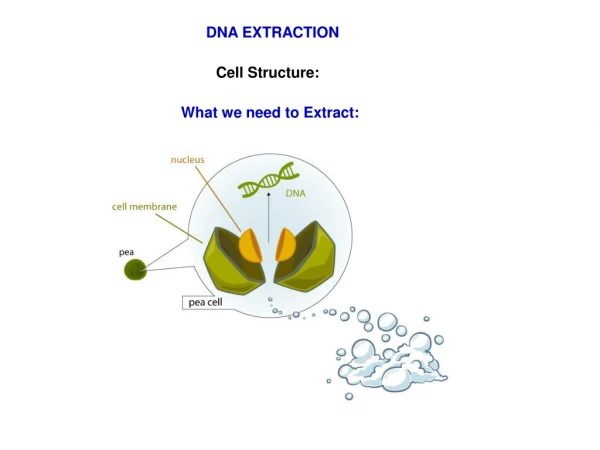
DNA EXTRACTION Cell Structure: What we need to Extract:
Extracting DNA at home 1/2 cup of split peas (100ml) 1/8 teaspoon table salt (less than 1ml) 1 cup cold water (200ml) Blend on high for 15 seconds. Strain through – Use Cheese Cloth Add 30ml (2 Tbl spoons) Detergent Mix and let sit for 5-10 mins. Transfer to test tubes Add Pinch of meat tenderizer
Precipitating the DNA Add an equal amount of Rubbing alcohol slowly down the side of the tube DNA will rise into the alcohol layer from the pea layer. use a wooden stick or other hook to draw the DNA into the alcohol.
Use of Ultrasound energy to break intermolecular interactions – Disrupts cell membrane releasing cellular contents into the media. What Principals were Involved? • Break open cells by grinding or sonication, • Remove membrane lipids by adding a detergent. • Remove cellular and histone proteins bound to the DNA, by adding a protease, or by i) Salt Precipitation using sodium or ammonium acetate, ii) using a phenol-chloroform extraction step. • Precipitate DNA in cold ethanol or isopropanol, DNA is insoluble in alcohol and clings together; this step also removes salt. • Wash the resulting DNA pellet with alcohol • Solubilize the DNA in a slightly alkaline buffer The DNA will dissolve in the aqueous layer, and everything else will go into the non-aqueous layer
Lysis of Cell Membrane – Use Detergent Hydrophilic Head Hydrophobic Tail Lipid Bilayer Micelle formation
Phenol Chloroform Extraction • Add one equal volume of phenol:chloroform to DNA solution. • Mix gently; spin for 5 minutes at max speed. • Carefully Remove aqueous layer (top layer – Avoid the Interface) to a new tube. Dump the bottom layer into a liquid waste container. • Add equal volume of chloroform. • Mix; spin 2 minutes at max speed. • Remove aqueous (top) layer to a new tube. Dump bottom layer into waste container. Note: Phenol is Dangerous – see MSDS – Should be handled in the hood
Salt Precipitation • This is the most common way to precipitate proteins • Protein Solubility is a function of • PhysicoChemical properties of the protein • pH • Temperature • Salt Conc. Use of Ammonium Acetate - high solubility and is relatively inexpensive At low conc. the solubility increases – Salting in At High conc. the solubility decreases – Salting out
DNA Pellet Ethanol Precipitation • Add sodium acetate to 0.3M - • Add two volumes 100% ethanol • Mix; spin 30 minutes at 4C • Wash: Remove supernatant carefully Fill tube halfway with 70% ethanol; spin 2 minutes. • Repeat wash. • Carefully pipette out or decant supernatant. There will be a clear pellet on the bottom. It may be difficult to see. • Dry the pellet by placing the tube upside down on a rack. It shouldn’t take longer than 30 minutes – just until all residual ethanol has evaporated. • Dissolve pellet in appropriate amount of TE or desired buffer
Plasmid DNA Extraction • Circular extra-chromosomal DNA molecule separate from the chromosomal DNA which is capable of replicating dependently of the chromosomal DNA • Codes for various genes e.g. Antibiotic Resistance • Plasmid DNA isolation requires separation of this DNA from • the chromosomal DNA in the bacterial cell • the polysaccharides, • lipids and • proteins that constitute the cell • ?????
Plasmid DNA Extraction • Cells are lysed by strong alkali (NaOH) and proteins are denatured by strong alkali and strong detergent (SDS) • The detergent complexes are then precipitated with a neutralizing salt (NH4oAC) • The plasmid is separated from the bacterial DNA by virtue of the Leaving the plasmid preparation in alkali for too long will destroy the plasmid DNA as well. plasmid's relative stability in alkali. • The chromosome will be precipitated by the salt and detergent. It is important, therefore, not to mix the solution to vigorously and release the chromosomal DNA from it trap • The plasmid is smaller and will remain free in solution. • The plasmid solution is separated from the cellular debris by centrifugation, • Further purification can be done by adhesion to powdered glass. Under high ionic strength (salty), the DNA binds to cationic glass and may be rinsed clean.
Plasmid DNA Extraction 1. Harvest the Bacterial cells - Spin 10 ml E.coli/plasmid suspension in 50 ml Falcon tube at 3000rpm for 10 minutes. 2. Carefully pour off supernatant without disturbing the cell pellet. Invert the tube and let it stand for 1 minute. 3.Resuspend cells in Buffer - Add 200ul of glucose/Tris/EDTA to the pellet and gently resuspend the pellet. 4. Transfer the entire suspension to a microfuge tube. 5. Lyse the cells Add 400ul of SDS/NaOH. Cap the tube and mix it very well. 6. Neutralize the alkali - the Add 300ul of KoAc. Mix well and keep on ice for 5 minutes. 7. Pellet Proteins and Chromosomal DNA - Spin tubes in microfuge at 12000rpm for 5-10 minutes. 8. Precipitate Plasmid - Transfer supernatant into another microfuge tube. Add equal volume of isopropanol. Close cap and mix well by rapidly inverting the tubes. 9. Leave at room temperature for 5 minutes. 10. Spin tubes at 12000rpm for 15 minutes. Pour off supernatant carefully. White pellet at the bottom of the tube contains plasmid DNA and RNA. 11. Wash the Plasmid DNA - Add 700ul 70% ethanol and wash the pellet by microfuging at 12000 rpm for 15 minutes. 12. Vacuum dry the pellets carefully. 13. Resuspend the pellet in 100ul TE pH8.0. 14. Take 10ul of the above plasmid add 2ulRNAase dye and incubate at 370C for 10 minutes. 15. Load entire sample in 0.8% agarose gel and eletrophorese at 75 V for 1 hr. 16. Stain in dilute ethidium bromide solution for ten minutes, then photograph with U.V. transillumination.
Plasmid Conformations • "Nicked Open-Circular" DNA has one strand cut. • "Relaxed Circular" DNA is fully intact with both strands uncut, but has been enzymatically "relaxed" (supercoils removed). You can model this by letting a twisted extension cord unwind and relax and then plugging it into itself. • "Linear" DNA has free ends, either because both strands have been cut, or because the DNA was linear in vivo. You can model this with an electrical extension cord that is not plugged into itself. • "Supercoiled" (or "Covalently Closed-Circular") DNA is fully intact with both strands uncut, and with a twist built in, resulting in a compact form. You can model this by twisting an extension cord and then plugging it into itself. • "Supercoiled Denatured" DNA is like supercoiled DNA, but has unpaired regions that make it slightly less compact; this can result from excessive alkalinity during plasmid preparation. You can model this by twisting a badly frayed extension cord and then plugging it into itself.
Plasmid Conformations • Usually 3 bands of Plasmid DNA • Highest - nicked or relaxed circular plasmidMiddle - linearLowest - supercoiled
Electrophoresis Migration of Charged Molecules under an electric field due to the electric surface charge of the molecules +Ve Anode Electric Field Strength -Ve Cathode
Migration Through the Gel If there is no hinderance Molecules would migrate at Almost the same rate Agarose is purified from agar, a gelatinous substance isolated from algae Agar: gelatinous substance chiefly used as a solid substrate to contain culture medium for microbiological work. It is an unbranched polysaccharide obtained from the cell membranes of some species of red algae,
1 2 3 4 5 DNA Ladder DNA Agarose Gel • Using DNA Ladder Can be used to estimate • Size –-Migration of the Bands • Quantity – Relative Intensities of the bands • Isolate particular (required) DNA band
UV DNA/RNA Quatitation The bases in DNA/RNA absorb UV light at Max at 260 nm 1 O.D. at 260 nm for double-stranded DNA = 50 ng/ul of dsDNA 1 O.D. at 260 nm for single-stranded DNA = 20-33 ng/ul of ssDNA 1 O.D. at 260 nm for RNA molecules = 40 ng/ul of RNA
Calculating the Amount of Sample For DNA - OD260 * 50 ng/ul * dilution factor For RNA - OD260 * 40 ng/ul * dilution factor Contamination Proteins Absorb at 280 nm thus ratio OD260/OD280 will give an indication of the purity of the sample preparation Pure preparations of DNA and RNA have OD260/OD280 values of 1.8 to 2.0, respectively Advantages of UV Quantitation: Does not chemically interfere with the sample Disadvantages: Impurities (e.g. nucleotides, RNA, EDTA and phenol) can also absorb at the given wavelengths
RNA Extraction High Quality RNA required for: Northern analysis, nuclease protection assays, RT-PCR, RNA mapping, in vitro translation and cDNA library construction. Problem: RNA is highly unstable – RNase! DEPC treated solutions – DiEthylPyroCarbonate 0.1% Autoclaved to yield CO2 and EtOH
RNA Extraction The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: - TRizol Reagent • Widely used for isolating total RNA from biological samples of different sources. • The principle - RNA is separated from DNA after extraction with an acidic solution containing guanidinium thiocyanate, sodium acetate, phenol and chloroform, Followed by centrifugation. • Under acidic conditions, total RNA remains in the upper aqueous phase, while most of DNA and proteins remain either in the interphase or in the lower organic phase. • Total RNA is then recovered by precipitation with isopropanol.
RNA Extraction • Pipet 300µl homogenized sample into clean epi tube. • Add 800µl Trizol LS. Mix by inverting. • Incubate 10 min at RT. • Add 0.2ml Chloroform. Mix vigorously by hand. • Incubate 15 min at RT. • Centrifuge 12,000g for 15 min, RT. • Transfer upper aqueous layer to a clean 1.5 ml epi tube. • Add 600µl Isopropanol to tube and invert several times. Incubate 10min, RT. • Centrifuge at 12,000 g for 10 min. • Pour off and discard supernatant. Wash pellet with 500µl 75% Ethanol. • Centrifuge 7500g for 5 min. • Pour off and discard supernatant. Look for pellet. Invert tube on paper towel and air dry pellet. • Resuspend pellet in 20-100µl MG H2O. • Perform Dnase I digestion on entire amount of RNA. Label should include Pure RNA. • Perform OD260/280 measurements and calculate the RNA concentration. • Use equal amounts of starting RNA template for all Reverse Transcription reactions.
mRNA Extraction • I. Prepare oligo-dT cellulose • use 40 mg oligo-dT cellulose / 1 mg total RNA • swell oligo dT-cellulose in elution buffer • wash oligo dT-cellulose 4 x with elution buffer (30 sec. full speed spin in between) • equilibrate oligo-dT cellulose with 2 to 3 washing steps using 1x binding buffer
II. mRNA purification • bring 1 mg total RNA with H2O to 600 µl • incubate 4 min at 65 °C • add 600 µl 2x binding buffer • add to 40 mg 1x binding buffer equilibrated oligo-dT cellulose • incubate 15 min at RT on a rolling incubator or vortex several times in between • spin oligo-dT cellulose down, discard supernatant • wash 2 x with 1x binding buffer • wash 2 x with wash buffer • elute with 250 µl elution buffer at 37 °C for 5 min • spin and keep supernatant (for second round of purification or precipitation) • elute oligo-dT cellulose again with 250 µl elution buffer • spin and keep supernatant (for second round of purification or precipitation) • combine eluate and add H2O to 600 µl • repeat mRNA purification by starting again with 4 min incubation at 65 °C mRNA Extraction
mRNA Extraction • III. Recover mRNA • add 50 µl 4 M NaCl and precipitate with 2 vol. cold EtOH • incubate 1 h at -20 °C • spin 10 min full speed, wash with 70 % EtOH, air dry and dissolve read A260 of 1 µl (100 to 250 fold diluted) • run 1 µg ( and 10 µg total RNA as comparison) on a 1.2 % agarose Formaldehyde gel
Lithium Chloride RNA Extraction • LiCl has been frequently used to precipitate RNA, although precipitation with alcohol and a monovalent cation such as sodium or ammonium ion is much more widely used. • LiCl precipitation offers major advantages over other RNA precipitation methods in that it does not efficiently precipitate DNA, protein or carbohydrate • It is the method of choice for removing inhibitors of translation or cDNA synthesis from RNA preparations • It also provides a simple rapid method for recovering RNA from in vitro transcription reactions.
mRNA Extraction Add an appropriate amount of oligo(dT) cellulose in binding buffer 1 to the RNA sample - allow to bind for 2 - 4 hours at room temperature in a total volume of 50 ml on a rotating wheel 8 To remove unbound (mainly ribosomal) RNA, spin the RNA - oligo(dT)cellulose mix at 5 000 rpm for 5 minutes. Decant the supernatant and resuspend in 25 ml of washing buffer warmed to 37oC. Mix on the rotating wheel for 15 minutes 9 Recover the RNA - oligo(dT)cellulose mix and wash again 25 ml of washing buffer warmed to 37oC. Mix on the rotating wheel for 15 minutes 10 Spin the RNA - oligo(dT)cellulose mix at 5 000 rpm for 5 minutes, decant the supernatant and recover bound poly(A+)RNA by eluting twice with 5 ml of 0.5% SDS in sterile water heated to 65oC. 11 Extract the eluate once with acid-phenol-chloroform. This removes any enzymatically active proteinase K and also any oligo(dT) cellulose that has been carried into the supernatant 12 Precipitate the RNA with one volume of isopropanol and 0.1 volumes of lithium chloride 2 and recover by centrifugation at 1200g for 10 minutes 13 Wash the pellet in 70% ethanol and then resuspend in 50 - 500 ml of 2 mM DTT, 1 u / ml RNasin in sterile Milli Q water 14 Quantitate by UV absorbance at 260 nm and check the ratio of UV absorbance at 260 and 280 nm