Download
1 / 25
250 likes | 534 Views
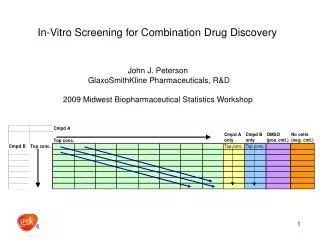
In-Vitro Screening for Combination Drug Discovery . John J. Peterson GlaxoSmithKline Pharmaceuticals, R&D 2009 Midwest Biopharmaceutical Statistics Workshop. Outline of Talk. Experimental design used What is “excess over highest single agent” (EOHSA)?

E N D
In-Vitro Screening for Combination Drug Discovery John J. Peterson GlaxoSmithKline Pharmaceuticals, R&D 2009 Midwest Biopharmaceutical Statistics Workshop
Outline of Talk • Experimental design used • What is “excess over highest single agent” (EOHSA)? • Simultaneous testing for EOHSA across all combinations • Some examples • An Introduction to “nonlinear blending” synergy.
Experimental design for screening pairs of compounds C = “combination” S = “single compound” V = “vehicle (control)” A kxk factorial. Here k = 9.
Excess over highest single agent (EOHSA) • For a kxk factorial design let be the mean response for the combination of • compound A at dose level i and compound B at dose level j. • Let be the dose of compound A alone at dose level i. • Let be the dose of compound B alone at dose level j. • The compound combination at dose levels (i, j) exhibits EOHSA if • EOHSA provides an easy-to-understand, low-level criterion for combination • drug screening. • - It is also an FDA criterion for (21 CRF 300.50) for combination drug approval. • - Used by CombinatoRx Inc. for combination compound screening (Borisy et al, • (2003) Proceedings of the National Academy of Science, 100, 7977–7982)
Simultaneous testing for EOHSA across compound combinations • Testing for EOHSA for one (i, j) compound combination can be accomplishedby use of the “min test” to test the null and alternative hypotheses below. • The above hypothesis can he tested by doing two one-sided tests of the form • each at level a, for an overall false-positive error rate of a (if both nulls are rejected) • However since there are combinations, we need to adjust for multiple comparisons to control the family-wise error rate across all combinations. • Hung (2000) and Westfall, Ho, and Prillaman (2001) have suggested procedures • for conducting simultaneous min tests.
Simultaneous testing for EOHSA across compound combinations • Since there are combinations, we need to adjust for multiplecomparisons to control the family-wise error rate across all combinations. • Hung (2000) and Westfall, Ho, and Prillaman (2001) suggested procedures for conducting simultaneous min tests. • However since the kxk design involves increasing doses of both compounds, • we can expect dose-response trends along each row and column of the design. • These dose-response trends can be exploited to gain additional power to testfor EOHSA across the various compound combinations.
Simultaneous testing for EOHSA across compound combinations • Suppose there is a trend forcompound A (for dose levels 0 to 5) • at dose level 4 of compound B. • Suppose also there is a trend forcompound B (for dose levels 0 to 4) • at dose level 5 of compound A. • It follows then that the compoundcombination (5,4) has EOHSA. C = “combination” S = “single compound” V = “vehicle (control)”
Simultaneous testing for EOHSA across compound combinations • So intersecting trends can be used test for EOHSA. • Since there are k dose levels of eachcompound there are 2(k-1) simultaneoustrend tests involving exactly l dose levels. • At each dose level, l, we can do 2(k-1)Bonferroni-adjusted trend tests. • The Tukey step-down trend test can be used as we step from level l=(k-1) to l=1. • The Tukey step-down test requires noadjustment of the (Bonferroni-adjusted) • a-level. C = “combination” S = “single compound” V = “vehicle (control)”
Simultaneous testing for EOHSA across compound combinations • PROPOSED TESTING PROCEDURE • 1. At each dose level, l, do 2(k-1)Bonferroni-adjusted trend tests. • 2. Use the Tukey step-down trend test as you step from level l=(k-1) to l=1. • Note: • The Tukey step-down test requires noadjustment of the (Bonferroni-adjusted) • a-level. • It can be proven that this procedurecontrols the FWER strongly at level a. • The proposed procedure is more efficientthan doing (k-1)2 multiple comparisons,even with correlation adjusted p-values. C = “combination” S = “single compound” V = “vehicle (control)”
Example • Mean percent reduction in living cancer • cells for 9x9 factorial experiment (n = 2) • replications per treatment group) for • compound A and compound B • (A549 cell line). • Means with boldface-italic font (in • the highlighted cells) are associated • with combinations having statistically • significant EOHSA.
Simultaneous testing for EOHSA across compound combinations • Proposed Bonferroni testing procedure: • 1. At each dose level, l, do 2(k-1)Bonferroni-adjusted trend tests. • 2. Use the Tukey step-down trend test as you step down from level l=(k-1) to l=1. • Proposed Modified Adjusted p-value testing procedure.In step 1 above replace the Bonferroni adjustment with a more efficient bootstrap adjusted p-value. • - This modification can be easily executed using SAS® PROC MULTTEST. - Simulations indicate that this procedure is more powerful than the Bonferroni adjusted version while still keeping the FWER to at most a.
6 It is possible to have many trends but no combinations with EOHSA So the null space ofthis testing procedure is complex. If the 5 above were replaced by the (circled) 6 then the combination highlighted in yellow would have EOHSA.
Simulations to assess overall Type I error rate • Consider the following three null hypothesis situations: Compound B only has a strong trend • 1,000 (normally distributed) data setswere simulated under each of the threenull hypotheses (with n=2 per cell ands = 1). Nominal a = 0.05. • The false-positive error rates were:
Comparisons with some other testing procedures Four drug combinations were tested in each of four cell lines resulting in 16 experiments MATBOOT=“multiplicity adjusted Tukey step-down trend test (Bootstrap)” MATBON=“multiplicity adjusted Tukey step-down trend test (Bonferroni)” SHUIIUT=Simes-Hommel Union-Intersection-Intersection-Union Trend SHUIIU=Simes-Hommel Union-Intersection-Intersection-Union MAPC=“multiplicity adjusted (bootstrap) p-values for (pairwise) contrasts.
Summary for Screening for EOHSA • Using the Bonferroni procedure to adjust Tukey’s step-down trend test results • in adjustments across only 2(k-1) groups rather than (k-1)2 groups. • - This provides improved power, even over Monte Carol adjusted procedures, • when their adjustment is over (k-1)2 groups (at least for k=9). • The power of this adjustment of Tukey’s trend test can be improved by using PROC MULTTEST, which is easy to implement. • It can be proven that the Bonferroni procedure to adjust Tukey’s step-down trend test • strongly controls the FWER. - The more powerful bootstrap adjusted modification appears to strongly control the FWER as well. • It may be useful to follow up this “low level” screening with a criterion that providesa higher “drug synergy hurdle”. One possibility is to use the concept of • “nonlinear blending” found in mixture experiments.
Nonlinear Blending Compared to FDA’s EOHSA. • Nonlinear blending is: “excess over highest single agent at total dose”as opposed to “excess over highest single agent (at component dose)” • Therefore nonlinear blending is a stronger form of “synergy”. drug B Total dose (molar) solid gray line of constant total dose (molar) drug A total dose (molar)
Point of 50% response Drug 2 d1+d2=A Isobologram (e.g. 50%) Drug 1 Why classical synergy indices do not generally work well for screening for combination drug synergy. We have Loewe synergy at thecombination (d1,d2) if But what if one or both of theED50’s (or ED60’s, etc.) do notexist?
Problems with the Interaction Index Cannot always compute the interaction index! Monotherapies do not achieve Y = 50% Yet, excellent synergy exists At a 50:50 ratio!
Combined compound response Compound B Total amount, T3 line of constant total amount, T3 Compound A total amount, T1 total amount, T3 total amount, T2 Nonlinear Blending. • If the response increases as we move away from the single agent compounds, then we have “optimal nonlinear blending”. • Optimal nonlinear blending can exist no matter what the shape of the dose response curves for the single agent compounds. • This type of synergy is much stronger than “excess over highest single agent”.
Response (percent) Response (percent) Strong nonlinear blending Weak nonlinear blending 100 100 75 75 50 50 25 25 0 0 0 50 100 0 50 100 Percent of drug 1 Percent of drug 1 Nonlinear Blending: Weak and Strong Nonlinear Blending Blending profiles for two different pairs of drugs at a given total (molar) dose For details see: Peterson, J. and Novick, S., “Nonlinear Blending: A Useful, General Concept for the Assessment of Combination Drug Synergy”, Journal of Receptors and Signal Transduction, vol.27, pp125-146.
Interpolated response 0 50 100 Percent of drug A Interpolated datapoints from platesA and B. The testing process for strong nonlinear blending. drug B(nM) 7 Rays (diagonals) of constant dose ratio. drug A (nM) Plate A = actual data point= interpolated data point
Ten ‘Total (Molar) Dose’ SlicesThrough the Combination Drug Region Drug B 99% 7 diagonals (rays) Interpolated response 50% This line only cuts across 4 true rays Imputed values in blue 50% plate A 1% plate B 0 50 100 Percent of drug A 0% 0% 50% 99% Drug A
Interpolated response 0 50 100 Percent of drug A Interpolated datapoints from platesA and B. The testing process for strong nonlinear blending. 1. For each total dose (molar) amount, fit a cubic polynomial curve to the data. (For robustness sake, we fit the data using a rank transformation of the percent drug A levels.) 2. Using gridding, find the maximum mean response. 3. Using the ‘min’ test, test to see if the maximum mean response is greater than both mean responses associated with 0% and 100% of drug A. If so, then we have strongnonlinear blending at that total (molar) amount. 4. Adjust the min test p-values (e.g. using the Hommel adjustment) across the various total dose levels used.
Summary for Screening for Strong Nonlinear Blending • Strong nonlinear blending can, in a practical sense, address any situation that • might come up in the screening of drug combinations. • The classical synergy indices such as Loewe synergy index, Chou & Talalay, Bliss, etc. all have serious flaws with regard to computation or interpretation ofsynergy. • If Loewe synergy can exist (e.g. both ED50’s exist) then the existence of strong nonlinear blending implies the existence of Loewe synergy. • It may be possible to improve screening for ‘strong nonlinear blending’ • by use of generalized additive models to automate fitting of response surfacesto 9x9 factorial plate designs. This is work for the future! For further details on Nonlinear Blending see: Peterson, J. and Novick, S., “Nonlinear Blending: A Useful, General Concept for the Assessment of Combination Drug Synergy”, Journal of Receptors and Signal Transduction, vol.27, pp125-146. Send e-mail to john.peterson@gsk.com for a copy.