Download
1 / 47
470 likes | 486 Views
Explore the potential of structural phylogenomic inference in predicting protein functions, including active site identification and subfamily specificity positions. Address existing errors in functional annotations and challenges in domain analysis for accurate predictions.

E N D
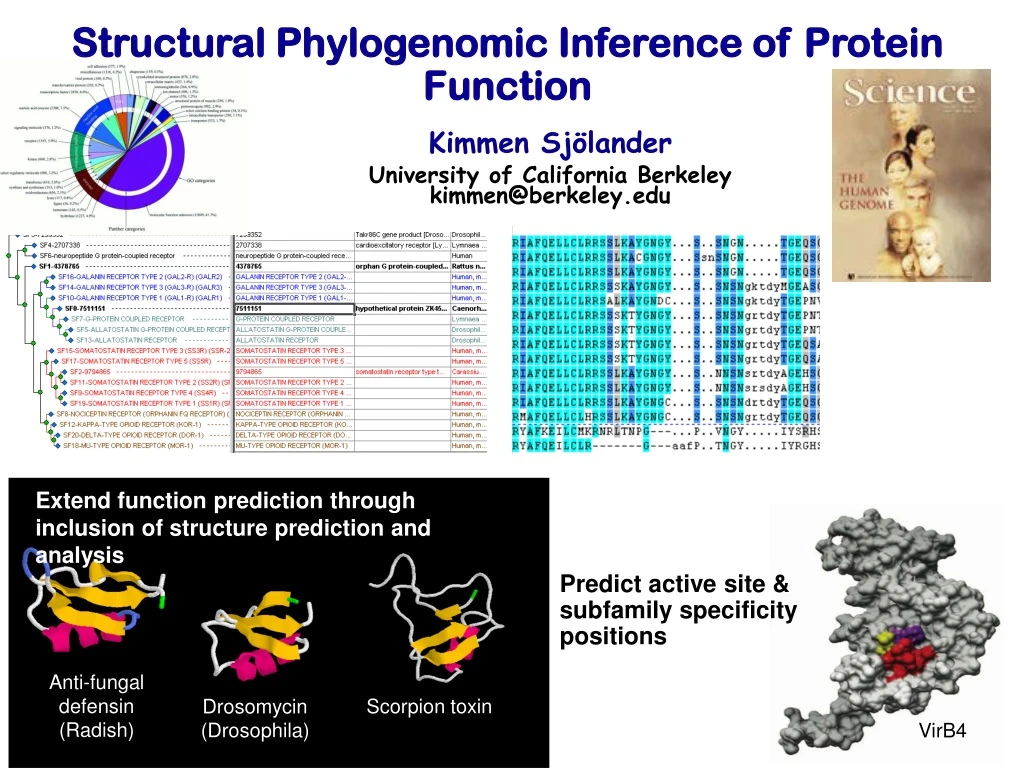
Structural Phylogenomic Inference of Protein Function Kimmen Sjölander University of California Berkeley kimmen@berkeley.edu Anti-fungal defensin (Radish) Scorpion toxin Extend function prediction through inclusion of structure prediction and analysis Predict active site & subfamily specificity positions Drosomycin (Drosophila) VirB4
Annotation transfer by homology • Status quo approach to protein function prediction • Given a gene (or protein) of unknown function • Run BLAST to find homologs • Identify the top BLAST hit(s) • If the score is significant, transfer the annotation • If resources permit, predict domains using PFAM or CDD • Problems: • Approach fails completely for ~30% of genes • Of those with annotations, only 3% have any supporting experimental evidence • 97% have had functions predicted by homology alone* • High error rate * Based on analysis of >300K proteins in the UniProt database
BLAST against Arabidopsis Panther PFAM results Tomato Cf-2 Bioinformatics Analysis Domain fusion and fission events complicate function prediction by homology, particularly for particularly common domains (e.g., LRR regions). Domain structure analysis (e.g., PFAM) is often critical. Tomato Cf-2 (GI:1587673) Dixon, Jones, Keddie, Thomas, Harrison and Jones JDG Cell (1996) Top BLAST hit in Arabidopsis is an RLK!
Error presumably due to non-orthology of database hits used for annotation
Phylogenetic analysis suggests it’s more likely a Biogenic Amine GPCR
Human neutral sphingomyelinase or bacterial isochorismate synthase?
Database annotation errors Main sources of annotation errors: Domain shuffling Gene duplication (failure to discriminate between orthologs and paralogs) Existing database annotation errors Propagation of existing database annotation errors Errors in gene structure Contamination Other… Galperin and Koonin, “Sources of systematic error in functional annotation of genomes: domain rearrangement, non-orthologous gene displacement and operon disruption.”In Silico Biol. 1998
Phylogenomic inference Eisen “Phylogenomics: Improving Functional Predictions for Uncharacterized Genes by Evolutionary Analysis,” Genome Research 1998 Sjölander, “Phylogenomic inference of protein molecular function: advances and challenges," Bioinformatics 2004
There is nothing more difficult to take in hand, more perilous to conduct, or more uncertain in its success, than to take the lead in the introduction of a new order of things. Because the innovator has for enemies all those who have done well under the old conditions, and lukewarm defenders in those who may do well under the new. This coolness arises partly from the incredulity of men, who do not readily believe in new things until they have had a long experience of them.
Include homologs from other species Construct multiple sequence alignment Construct phylogenetic trees. Overlay with annotation data. Identify subfamilies. Retrieve key literature Construct HMMs for the family and for individual subfamilies. Predict protein structure Predict key residues Predict cellular localization. Deposit book in library Construction of genome-scale phylogenomic libraries Cluster genome into global homology groups
Berkeley Universal Proteome Phylogenomic Explorer 9,707 protein family “books” and 708K HMMs and expanding daily http://phylogenomics.berkeley.edu/UniversalProteome
Protein fold prediction 12% identity VirB4 TrwB structure (1E9RA) Active site
SCI-PHY subfamilies supported by ML tree, and also consistent with subtype and phylogenetic distribution (only one branch of ML tree displayed)
Database queries Look up protein family “books” based on the annotations associated with any sequence. Queries can be based on GO biological process, PFAM domains, UniProt accession numbers, etc.
Key algorithms in PhyloFacts library construction What clustering methods are appropriate for inference of protein function? What alignment methods are accurate? How to mask? What tree methods to use? How to root a tree? Can we define functional subfamilies automatically?
Fraction superposable positions drops with evolutionary divergence
FlowerPower Clustering global (or glocal) homologs Minimize profile drift Improved alignment accuracy Nandini Krishnamurthy, Ph.D.
Step 1: Construct SearchDB Q=query Construct SearchDB using PSI-BLAST against target database Q
Step 2: Select and align core set. Q Inclusion criteria: E-value 1e-10 Bi-directional coverage MUSCLE multiple alignment (Edgar, 2003)
Step 3: Run SCI-PHY to identify subfamilies and build subfamily HMMs (SHMMs) Q BETE subfamily identification: Sjölander 1998 SHMM construction: Brown et al, 2004
Step 4: SHMMs compete for sequences from SearchDB. Sequences meeting criteria are aligned to their closest SHMM. Q
Step 5: Run SCI-PHY on extended alignment to identify new subfamilies and construct SHMMs. Q
Comparing FlowerPower, BLAST, PSI-BLAST and UCSC T2KTest: Clustering global homologs Agreement at domain structure determined by PFAM. SCOP used to cluster PFAM domains into structural equivalence classes.
Seq1 LERY-K Seq2 LDRFPR Seq3 IERYGK Seq4 MDRF-K Seq5 VERYGK 5 3 1 4 2 Phylogenetic tree & subfamily decomposition Multiple sequence alignment Subfamily Classification In PHYlogenomics (SCI-PHY) Nandini Krishnamurthy, Ph.D. Duncan Brown Agglomerative clustering Input: MSA Initialize: construct profile1 for each row in MSA While (#clusters > 1) { Join closest2 pair of clusters Re-estimate profile1 Compute encoding cost3 for this stage } /* cut tree using minimum encoding cost */ Use Dirichlet mixture densities Distance function: relative entropy Detection of critical positions Sjolander, K. "Phylogenetic inference in protein superfamilies: Analysis of SH2 domains" Proceedings of Conference Intelligent Systems for Molecular Biology (ISMB) 1998
Cost N 1 # classes Subfamilies identified using minimum encoding cost principles • Each stage of the algorithm defines a different set of alignments, one for each cluster (“subfamily”). • Find the point during the clustering where the encoding cost of the alignments is minimal. This defines the subfamily decomposition. N= number of sequences. S= number of subfamilies; n c,1…n c,s are the amino acids aligned by subfamilies 1 through s at column c. represents the Dirichlet mixture prior.
SCI-PHY analysis of selected GPCRs Venter et al, The sequence of the human genome (2001) Science. Sjolander, “"Phylogenomic inference of protein molecular function: advances and challenges," (2004) Bioinformatics
Y221 W222 D558 R627 H745 D628 Y743 A744 G629 Key residue prediction using subfamily and family-wide conservation analysis Elizabeth Hua-Mei Kellogg Ryan Ritterson Nandini Krishnamurthy Parker JS, Roe SM, Barford D. , EMBO J., 2004 Tanaka Hall, T. Structure 2005 Rivas et al, 2005 D RD E YAH
3.5.2.2 Dihydropyrimidinase 3.5.4.1 Cytosine deaminase 3.5.2.3 Dihydroorotase 3.5.1.5 Urease Subfamily Function Prediction Using HMMs 7TM GPCR ABC Transporter Amidohydrolase ATPase Family
3 4 5 1 2 6 7 • At completely conserved positions, and subfamily gapped positions: Use match state distributions estimated for general (family) HMM. • At other positions: • Estimate Dirichlet mixture density posterior for each subfamily at each position separately. • Use Dirichlet density posteriors to weight contributions from other subfamilies. • Compute amino acid distribution using weighted counts and standard Dirichlet procedure. Subfamily HMM construction Error Brown et al,“Subfamily HMMs in functional genomics” (2005) Pacific Symposium on Biocomputing
Subfamily HMMs increase the separation between true and false positives 1.5% error rate in subfamily classification using top-scoring SHMM • 515 unique SCOP folds • PFAM full MSAs • Scored against Astral PDB90
SATCHMO: Simultaneous Alignment and Tree Construction using Hidden Markov mOdels Xia Jiang Nandini Krishnamurthy Duncan Brown Michael Tung Jake Gunn-Glanville Bob Edgar Edgar, R., and Sjölander, K., "SATCHMO: Sequence Alignment and Tree Construction using Hidden Markov models," Bioinformatics. 2003 Jul 22;19(11):1404-11
SATCHMO motivation • Structural divergence within a superfamily means that… • Multiple sequence alignment (MSA) is hard • Alignable positions varies according to degree of divergence • Current MSA methods not designed to handle this variability • Assume globally alignable, all columns (e.g. ClustalW)… • Over-aligns, i.e. aligns regions that are not superposable • …or identify and align only highly conserved positions (e.g., SAM software with HMM “surgery”) • Challenge • Different degrees of alignability in different sequence pairs, different regions • Masking protocols are lossy: loop regions may be variable across the family but may be critical for function!
SATCHMO algorithm • Input: unaligned sequences • Initialize: a profile HMM is constructed for each sequence. • While (#clusters > 1) { • Use profile-profile scoring to select clusters to join • Align clusters to each other, keeping columns fixed • Analyze joint MSA to predict which positions appear to be structurally similar; these are retained, the remainder are masked. • Construct a profile HMM for the new masked MSA } • Output: Tree and MSA
Assessing sequence alignment with respect to structural alignment Xia Jiang Duncan Brown Nandini Krishnamurthy
Catalytic residues colored red Future work: Interactive specificity position identification • Enable users to select subtrees for analysis • Identify positions conserved within each subtree, but which differentiate the two** • Plot over MSA and on structure (if available) Donald and Shakhnovich, NAR 2005
Major challenge: Phylogenetic uncertaintyGiven: A (gene tree of unknown function), gene trees B and C (characterized function)Predict function for A. A B B A C C A C B Problem: use three phylogenetic tree methods, get 3 or more trees! Change the MSA, you also change the tree… Need: Better simulation studies, benchmark datasets
http://phylogenomics.berkeley.edu Berkeley Phylogenomics Group PI: Kimmen Sjölander Nandini Krishnamurthy, Ph.D. Duncan Brown Sriram Sankararaman Xia Jiang Jake Gunn-Glanville Lead programmer and web administrator: Dan Kirshner This work is supported in part by a Presidential Early Career Award for Scientists and Engineers from the NSF, and by an R01 from the NHGRI (NIH).