Download

1 / 63
640 likes | 1.31k Views
Structure of Chromosomes and Microbial Growth Eustace Smith Objectives To give an overview of the structure of bacterial chromosomes To describe the growth of microorganisms and the influence of environmental factors Learning Outcomes

E N D
Structure of Chromosomes and Microbial Growth Eustace Smith
Objectives • To give an overview of the structure of bacterial chromosomes • To describe the growth of microorganisms and the influence of environmental factors
Learning Outcomes • At the end of this section, students will have a good understanding of the following • The structure of bacterial chromosomes and plasmids • DNA super-coiling and the functions of topoisomerases • The process of growth in bacteria • a. growth curve (cycle) • b. growth in suspension versus growth in colony
The methods available for measuring microbial growth • a. Total and viable counts • b. Cell mass determination • c. Turbidity measurements • d. Serial dilution of cell suspensions • Chemostat versus batch culture
Environmental effects on microbial growth (and types of microorganisms) • a. Temperature • b. Oxygen • c. pH • d. Osmotic effects (water activity)
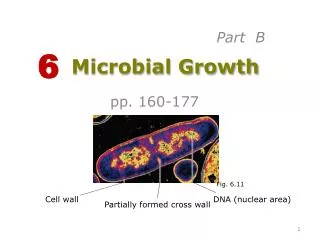
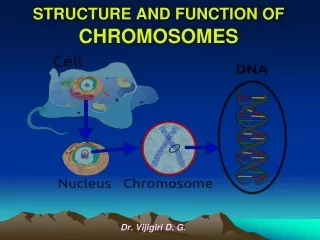
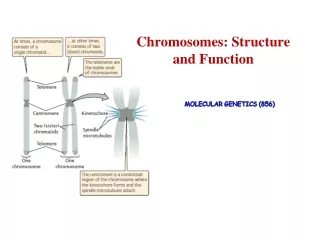
Chromosomal Structure • The bacterial chromosome is usually a covalently closed circular molecule • There are some bacteria hwvr that have linear chromosomes e.g. Borrelia burgdoferi • Plasmids which are small circles which contain extra-chromosomal DNA also exist • In E. coli, the chromosomes contains @ 4,600 Kb
This DNA is twisted and folded giving a super coiled structure • The super coiled structure is further organized into super coiled domains • There are over 50 super coiled domains in E. coli • These super coiled domains are stabilized by association with structural proteins
DNA Structure: Supercoiling • Super coiling is the state in which double stranded DNA molecules are further twisted • Super coiling puts the DNA molecule under torsion. This torsion is held only if the circular structure is maintained • Negative super coiling occurs when DNA is twisted about its axis in the opposite direction from a right handed double helix
It is in this form that super coiled DNA is predominantly found in nature.
Topoisomerases • In Bacteria & the Archae, there is a special enzyme Topoisomerase 11 (DNA gyrase) which introduces negative super coils • The process of super coiling has several stages • A. The DNA molecule is twisted • B. A break occurs where the two chains come together.
Introduction of supercoiling in circular DNA by Topoisomerase 11
c. This Break is brought about by Topoisomerase 11 • d. The broken double helix is resealed on the opposite side of the intact strand • e. This results in super coiled DNA
Topoisomerase 1 • A topoisomerase is defined as an enzyme that affects the Topology of DNA. Topology is the branch of mathematics that deals with the properties of geometric figures that are unaltered when the figures are twisted or contorted • There is another enzyme that is able to remove the super coiling in DNA. This is called topoisomerase 1.
Topoisomerase 1, introduces a single- strand break in the DNA and causes the rotation of one single strand of the double helix around the other • A nick (break in the backbone) of either strand allows the DNA to return to the relaxed state • Because the bacterial chromosome contains ~50 super coiled domains, a nick in the DNA of one of these does not relax the DNA in the others.
Through the action of Topoisomerase, the DNA molecule can be alternatively super coiled and relaxed • Super coiling is necc for packing DNA into the confines of a cell • Relaxing is necc to allow DNA to be replicated • Super coiling also affects gene expression
Some genes are more actively transcribed when DNA is super coiled • Transcription of others is inhibited by excessive supercoiling
The Process of growth in Bacteria • Definitions • a. Growth – An increase in the number of cells • b. Growth rate – The change in cell number or cell mass per unit time • c. Generation Time – The interval for the formation of two cells from one
A typical bacterial growth curve is divided into the following • a. Lag phase • b. Exponential phase • c. Stationary phase • d. Death
Lag Phase • Usually when a microbial population is inoculated into fresh medium, there is a brief or extended time period before growth begins • This is called the lag period • The length of the lag period depends on the history of the culture and the growing conditions.
a. If an exponentially growing culture is inoculated to fresh medium under the same conditions, a lag is not seen but growth begins immediately • b. If the inoculum is from an old culture (stationary phase), a lag occurs even if all the cells are viable. This is because these cells are usually depleted of various essential constituents and require time to resynthesize these
c. A lag also ensues when cells have been damaged e.g. by radiation, heat or toxic chemicals. This because time is required to repair the damage • d. A lag also occurs when a population is transferred from a rich culture medium to a poorer one. For growth to occur in the poorer culture medium the cells must have a complete complement of enzymes for synthesis of the essential metabolites not present in that medium. The time is thus required for the synthesis of the new enzymes
Exponential Phase • Exponential growth – This is the growth of a microorganism where the cell number doubles within a fixed time period. • When one plots the cell numbers of an exponentially growing culture against time on arithmetic coordinates one gets a curve with a constantly increasing slope. If one were to plot the log number of cells versus time (on semi log paper). A straight line is the result.
Figure 6 • One of the characteristics of exponential growth is that the rate of increase in cell number is slow initially but increases at an ever increasing rate. • A practical implication of this is that when a non sterile product like milk is allowed to stand under ideal conditions of growth, the early stages of growth are not detrimental but doubling or tripling this time results in spoilage.
Plot of cell number vs time for a population that doubles every 30 min
Stationary Phase • In a batch culture exponential growth cannot occur indefinitely. After a while: • a. An essential nutrient from the culture medium is used up and/or • b. Some waste product of the organism builds up in the medium • At this stage the population has reached the stationary phase
Definition of Stationary Phase – the period immediately following exponential growth when the growth rate of the population falls to zero • In the stationary phase no net increase or decrease in cell number occurs. Many cell functions hwvr may still continue such as energy metabolism and some biosynthetic processes. In some microorganisms slow growth may occur accompanied by the death of some cells resulting in no net change in cell number. This is called cryptic growth
Death Phase • If incubation continues after a population reaches the stationary phase, the cells may remain alive & continue to metabolize or they may die. When they die the population is said to be in the death phase. In some cases death may be accompanied by cell lysis, where there is loss of cellular integrity and the release of cytoplasmic contents.
Growth on solid Media or in suspension • Microorganisms may grow in cell suspension – broth cultures or on agar medium – plate cultures • Growth in cell suspension is accompanied by increased turbidity • Growth on solid medium – On solid medium one cell or a group of cells continue to multiply until they become visible as a colony of microbial cell
Methods for measuring Microbial Growth • Total Cell Count - The number of cells in a population can be measured by counting a sample under the microscope. This is called a direct microscopic count • Two kinds of direct microscopic counts are done • a. On samples dried on slides • b. On samples in liquid • With liquid samples special counting chambers are used.
Direct microscope count with Petroff-Hausser counting chamber
How to calculate cell number using Petroff-Hausser counting chamber • All cells in a large square are counted • The number of cells in several large squares are averaged • To calculate the number of cells/ml of sample you multiply the average cell number by the number of squares (25) x 1/0.02 x 1000.
Breakdown of calculation • The space between the coverslip and the slide is 0.02 mm. • The entire grid has 25 large cells, and a total area of 1 mm2. • The total volume is thus 0.02 mm x 1 mm2 = 0.02 mm3 • 1 ml is 1 cm3, 1cm3 is 1000 mm3 • Each grid thus has a volume of 0.02/1000 ml
If each grid had 100 cells the one ml would have 100 x 1000/0.02 cells = 100 x 50,000 or 5,000,000. • Each grid has 25 large squares, if each square had an average of 10 cells then the grid would have 250 cells. One ml would then have 250 x 50,000 cells = 12,500,000
Limitations of direct microscopic counting • Direct microscopic counting is a quick way of estimating microbial cell number. Hwvr it has several limitations • 1. Dead cells are not distinguished from living cells • 2. Small cells are difficult to see under the microscope & some will likely be missed • 3. Precision is hard to achieve
4.A phase contrast microscope is required when the sample is not stained • 5. Method is not suitable for cell suspensions with less than 106/ml. Here very few bacteria will be seen in the microscope field. Here one would have to first concentrate cells and re-suspend in a small volume.
Viable Count • This method counts only live or viable cells • A viable cell is defined as one that is able to divide and form offspring • Viable counts usually determine the number of cell in a sample capable of forming colonies on a suitable agar medium • The viable count is thus often called the plate count or colony count. The assumption made here is that each viable cell can yield one colony
There are two ways of performing a plate count • a. The spread plate method • b. Pour plate method
Spread Plate Method • A volume of an appropriately diluted culture usually ~0.1 ml is plated over the surface of an agar plate using a sterile glass spreader • The plate is incubated under appropriate conditions
Pour Plate Method • A known volume of appropriately diluted culture usually (0.1-1.0 ml) of culture is pipetted into a sterile petri plate • Cooled molten agar medium is added • Petri plate contents are gently swirled for mixing • Incubated under appropriate conditions • The colonies that appear are counted
Serial Dilution of cell suspensions • With pour and spread plate methods it is important that the plates to be counted are neither too sparse nor overcrowded • If a plate is inoculated with too many cells, some colonies may not form or others may fuse • If there are too few colonies the counts will not be statistically valid • In practice only plates with between 30-300 colonies are counted
To obtain the appropriate colony number the sample is usually dilute • Serial ten fold dilutions are usually used for plating with those plates having between 30-300 colonies counted • A 10 fold (10-1) dilution can be achieved by mixing 1 ml of sample with 9 ml of diluent • A 100 fold (10-2) dilution can be achieved by mixing 1 ml of sample with 99 ml of diluent
Alternatively a 100 fold dilution can be achieved by making successive 10 fold dilutions • A 10-6 dilution can be obtained by performing six consecutive 10 fold dilutions and so on