Download
1 / 1
10 likes | 113 Views
This study focuses on accurate modeling of Krn+ clusters' electronic states and potential energy surfaces using ab initio and DIM methods to analyze rovibrational spectra. It involves sophisticated calculations and incorporation of spin-orbit coupling for reliable results.

E N D
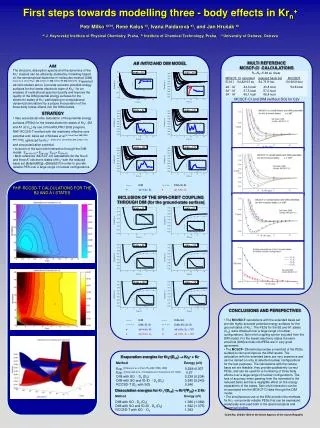
First steps towards modelling three - body effects in Krn+ Petr Milko a) b), René Kalus c), Ivana Paidarová a), and Jan Hrušáka) a) J. Heyrovský Institute of Physical Chemistry, Praha, b) Institute of Chemical Technology, Praha, c) University of Ostrava, Ostrava AB INITIO AND DIM MODEL MULTI REFERENCE MCSCF-CI CALCULATIONS R1=R2=5.38 au linear MCSCF- CIextendedreduced basis set RCCSDT E(1A’) -54.82103 au -54.76111au -54.92414au 2A´-1A´ 44,0 kcal 43,8 kcal 54.8 kcal 3A´-1A´ 57,5 kcal 57,0 kcal 5A´-1A´ 66,1 kcal 66,4 kcal MCSCF-CI and DIM (without SO) for C2v INCLUSION OF THE SPIN-ORBIT COUPLING THROUGH DIM (for the ground-state surface) • AIM • The structure, absorption spectra and the dynamics of the Krn+ clusters can be efficiently studied by modelling based on the semiempirical diatomics-in-molecules method (DIM) [Kalus et al, Chem.Phys. 294 (2003)141; 298 (2004)155;302(2004)279]. Presented ab initio studies aim to i) provide accurate potential energy surfaces for the lowest electronic state of Kr3+ for an analysis of rovibrational spectra ii) justify and improve the quality of the DIM potential energy surfaces for the electronic states of Krn+ participating in computational dynamical simulations by a proper incorporation of the three-body forces effects into the DIM models. • STRATEGY • Very accurate ab initio calculation of the potential energy surfaces (PESs) for the lowest electronic states of Kr3+ (B2 and A1 at C2v,) by use of the MOLPRO 2002 program, RHF-RCCSD-T method with the relativistic effective core potential and basis set of Niclass et al [J.Chem.Phys.102(1995) 8942 Dolg]] optimized for Kr2+ [Kalus at al, Chem.Phys.294 (2003) 141] • and core polarization potential. • Inclusion of the spin-orbit interaction through the DIM model: Eab initio SO= Eab initio- EDIM+ EDIM+SO • Multi-reference (MCSCF-CI) calculations for the five A’ and three A’’ electronic states of Kr3+ with the reduced basis set [8s6p6d6f2g]->[8s6p3d1f] in order to provide reliable PES over a large range of nuclear configurations. RHF-RCCSD-T CALCULATIONS FOR THE B2 AND A1 STATES • CONCLUSIONS AND PERSPECTIVES • The RCCSD-T calculations with the extended basis set provide highly accurate potential energy surfaces for the ground states of Kr3+. The PESs for the B2 and A1 states (C2v)were obtained over a large range of nuclear configurations. Spin-orbit coupling can be included from the DIM model. For the lowest electronic states the semi-empirical (DIM) and ab initioPESs are in very good agreement. • The MCSCF- CI method provides a manifold of the PESs suitable to test and improve the DIM results. The calculation with the extended basis are very expensive and can be carried on only at selected nuclear configurations for the test purposes. The calculations with the reduce basis set are feasible, they provide qualitatively correct PESs, and can be used for a monitoring of three body effects over a large range of nuclear configurations. The loss of accuracy when passing from the extended to the reduced basis set has a negligible effect on the energy separations of the states. Spin-orbit interaction can be incorporated into the MCSCF-CI data through the DIM model. • The simultaneous use of the DIM and ab initio methods for Kr3+ can provide reliable PESs that can be expressed analytically and used both in the spectroscopical and dynamical studies. Evaporation energies for Kr3+(Dh) Kr2+ + Kr MethodEnergy (eV) Exp. [Hiraoka et al, J.Chem.Phys.92 (1990) 4408] 0.2290.007 Exp. [Fehsenfeld et al, 31st Gaseous El. Conference, NY 1978] 0.27 DIM with SO - De (D0) 0.238 (0.234) DIM with SO and ID-ID - De (D0) 0.245 (0.240) RCCSD-T (De with SO) 0.246 Dissociation energies forKr3+(Dh) Kr+(2P3/2) + 2 Kr MethodEnergy (eV) DIM with SO - De (D0) 1.386 (1.369) DIM with SO and ID-ID - De (D0) 1.392 (1.375) RCCSD-T with SO - De 1.393 Grant No. 203/02/1204 of the Grant Agency of the Czech Republic