Download

1 / 82
820 likes | 1.2k Views
Protein Classification and Meta-organization. Methods for Global Organization of the Protein Universe. Golan Yona. Department of Computer Science Cornell University. Ismb02. Outline. Introduction Sequence-based classifications of proteins Methods Sequence comparison algorithms

E N D
Protein Classification and Meta-organization.Methods for Global Organization of the Protein Universe Golan Yona Department of Computer Science Cornell University
Ismb02 Outline • Introduction • Sequence-based classifications of proteins • Methods • Sequence comparison algorithms • Mathematical models of protein families • Databases • Domain-based protein sequence classification • Protein-wise protein sequence classification • Alternative representations of proteins • Structure-based classifications of proteins • Structure comparison algorithms • Databases • Other classifications
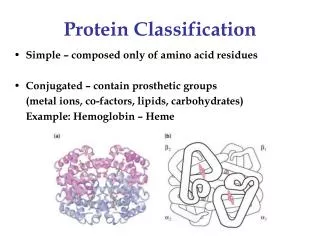
C M A L V L E C V Ismb02 What are proteins ? • Structural framework (keratin, collagen) • Transport and storage of small molecules (hemoglobin) • Transmit information (hormones, receptors) • Antibodies • Blood clotting factors • EnzymesThe protein is created in the cell as a unique sequence • of amino acids
Sequence ACMVLLCEVEKYP… folding Structure Function ????? Ismb02
Ismb02 Background and Problem definition About protein sequences are known today (non-redundant database). This number keeps rapidly growing (large scale sequencing projects). ! The function of 40-50% of the new proteins is unknown. Understanding biological function is important for: • Study of fundamental biological processes • Drug design • Genetic engineering
Ismb02 Basic approach • Calculate pairwise distances/similarities with sequences of known proteins • Pairwise similarities are computed using the rigorous dynamic programming algorithm (Smith-Waterman), or heuristic algorithms such as Fasta and Blast. V MCTKKLLVYD VRSM KLLLGF E
Database search • A sequence is compared with all sequences in the database • Its properties are extrapolated from neighboring sequences (nearest neighbor approach to learning and generalization) MFGPVRACAATFD... MCTKKLVTD.... RVSMLILLGFH..... KAACGCGLMDRFYVVLVV.... .....
BLASTP 2.2.2 [Jan-08-2002] Reference: Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schaffer, Jinghui Zhang, Zheng Zhang, Webb Miller, and David J. Lipman (1997), "Gapped BLAST and PSI-BLAST: a new generation of protein database search programs", Nucleic Acids Res. 25:3389-3402. Query= nr|001640000880 trembl: (Q935W2) DNA mismatch repair protein (Fragment). (164 letters) Database: /local/databases/nr/nr 933,075 sequences; 292,319,403 total letters Searching..................................................done Score E Sequences producing significant alignments: (bits) Value nr|008510000010 swissprot: (Q99XL8) DNA mismatch repair protein ... 316 5e-86 nr|001640000880 trembl: (Q935W2) DNA mismatch repair protein (Fr... 298 1e-80 nr|001640000882 trembl: (Q936C5) DNA mismatch repair protein (Fr... 290 3e-78 nr|001640000877 trembl: (Q933P8) DNA mismatch repair protein (Fr... 283 4e-76 nr|001640000883 trembl: (Q936C9) DNA mismatch repair protein (Fr... 280 5e-75 nr|001350001337 trembl: (Q9ETZ4) DNA mismatch repair protein (Fr... 248 2e-65 nr|001350001345 trembl: (Q9EVZ2) DNA mismatch repair protein (Fr... 247 3e-65 nr|001350000805 trembl: (Q933M4) Putative DNA mismatch repair en... 246 7e-65
BLASTP 2.2.2 [Jan-08-2002] Reference: Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schaffer, Jinghui Zhang, Zheng Zhang, Webb Miller, and David J. Lipman (1997), "Gapped BLAST and PSI-BLAST: a new generation of protein database search programs", Nucleic Acids Res. 25:3389-3402. Query= nr|001640000690 trembl: (Q22102) Hypothetical 19.0 kDa protein. (164 letters) Database: /local/databases/nr/nr 933,075 sequences; 292,319,403 total letters Searching..................................................done Score E Sequences producing significant alignments: (bits) Value nr|001640000690 trembl: (Q22102) Hypothetical 19.0 kDa protein. 310 5e-84 nr|009250000047 trembl: (Q9GUG1) Hypothetical 104.9 kDa protein. 230 4e-60 nr|009660000030 trembl: (Q20175) Hypothetical 110.0 kDa protein. 225 1e-58 nr|008350000033 trembl: (Q17506) Hypothetical 95.8 kDa protein. 152 8e-37 nr|011390000015 trembl: (O76601) H02F09.4 protein. 152 1e-36 nr|002650001251 trembl: (Q9XUR0) T05F1.12 protein. 148 2e-35 nr|002140000359 trembl: (O16440) Hypothetical 24.8 kDa protein. 139 2e-32 nr|002210001099 trembl: (Q9N5M4) Hypothetical 25.9 kDa protein. 135 1e-31
BLASTP 2.2.2 [Jan-08-2002] Query= nr|001640000685 trembl: (Q19708) F22B5.4 protein. (164 letters) Database: /local/databases/nr/nr 933,075 sequences; 292,319,403 total letters Searching..................................................done Score E Sequences producing significant alignments: (bits) Value nr|001670000430 trembl: (P90860) F36A2.7 protein. 153 8e-37 >nr|001670000430 trembl: (P90860) F36A2.7 protein. Length = 167 Score = 153 bits (386), Expect = 8e-37 Identities = 83/157 (52%), Positives = 108/157 (67%), Gaps = 2/157 (1%) Query: 8 RKTMFRFVLSRNASTSNVPSPARIQLKKPAEAGHFQYSRNWSRDPRFVKVAIQKGDTPYQ 67 + T+ R VL RNAS S + +K + G F+Y R+ SRD R+ A + GDT + Sbjct: 13 KNTIARIVLVRNAS-SGLTLKHEQTIKINDQQGFFKYQRDVSRDTRYSNPA-KPGDTAAR 70 Query: 68 FLVRRLGHAYEVYPLFVLTAAWFVLFCSASYWSFGKAEIWLDRSNSKAPWDWERLRDTYW 127 F+ R+LGHAYE+YPLF L A W VLF ++SF KAEIWLDRS++ APWDWER+R+ YW Sbjct: 71 FMFRKLGHAYEIYPLFGLLAIWCVLFGYTVWYSFEKAEIWLDRSHTVAPWDWERIRNNYW 130
Smith Waterman (Adv. App. Math 1981) Based on the dynamic programming algorithm. Finds the maximal local similarity given the parameters for point mutations s(ai,bj) and the gap penalties g The optimal alignment of the a1a2a3….aiwithb1b2b3….bj must end in one of three possible ways ai ORai OR- bj -bj If S(i,j) is the best score over all possible alignments of a1a2a3….aiwithb1b2b3….bj then S(i,j) = MAX{S(i-1,j-1)+s(ai,bj), S(i-1,j)-g, S(i,j-1)-g}
Smith Waterman (Adv. App. Math 1981) Implementation: 0 . . . j . . m 0 . i . n S(i-1,j-1)+s(ai,bj) S(i-1,j)-g S(i,j-1)-g
FASTA (Pearson & Lipman, PNAS 1988) Create a hash table with all k-tuples that appear in the query sequence, index vectors and an offset vector. Run a bounded dynamic programming around the best diagonal/region Library sequence 0 . . . j . . m 0 . i . n
BLAST and Gapped-BLAST (Altschul et al., JMB 1990, Altschul et al. NAR 1997) For each word length k in the query generate all k-tuples with score > T. Use these k-tuples to search the library sequence. Extend the seeds to generate HSPs (high scoring segment pairs without gaps). Combine HSPs 0 . . . j . . m 0 . i . n
p1 p2 … pn PSI-BLAST (Altschul et al. NAR 1997) • Iterative procedure that uses a position-specific scoring matrix. • Generate a profile P = p1p2…pn from hits detected at iteration I, to search the database in iteration I+1, until convergence. • The profile stores the probabilities to observe the 20 amino acids at each position along the query sequence (derived from hits with related sequences, after weighting and addition of pseudo-counts) 1 2 n 1 2 . . 20
Limitations of sequence comparison In many cases sequences have diverged to the extent that their sequence similarity is undetectable with the above algorithms. More than 50% of the 1,000,000 proteins that are known today have an unidentified function Iterative procedures such as PSI-BLAST are more powerful, but are more susceptible to parameter tuning and have higher false positive rate Use reliable mathematical models of protein families to classify new genes
Protein classification More specific Sub-families (rab, ras, ran, rac, rho, ARF1, transducin,..) Families (G-proteins, Motor proteins, DNA helicases,..) Super-families (P-loop containing nucleotide triphosphate hydrolases) Fold families (P-loop containing hydrolases, beta/alpha tim barrel) Classes (alpha and beta, all-alpha, ...) Common evolutionary origin
MCTKKLVTD.... Why is protein classification important? • Protein classification can help to elucidate the function of new genes. • Comparing a sequence with a database of protein families is more effective than a standard database search. • Each family is represented as a model • regular expression • profile • HMM • Those models can capture subtle similarities. Protein classification is useful in structure and function prediction, and especially important in large-scale annotation efforts. Family1 Family2 Family3
Databases of protein families • Domain-based clusterings: • Prosite (Bairoch & Bucher ISMB 1994, Laurent et al. NAR 2002). • Pfam (Sonnhammer et al. Proteins 1997, Bateman et al. NAR 2002). • ProDom (Gouzy et al. Computers and Chemistry 1999, Corpet et al. NAR 2000). • Prints (Attwood et al. NAR 2002). • Domo (Gracy & Argos Bioinformatics 1998). • Blocks (Henikoff et al. NAR 1999). • Protein-based clusterings: • ProtoMap (Yona et at. Proteins 1999, Yona et al. NAR 2000). • Cogs (Tatusov et al. Science 1997, Tatusov et al. NAR 2001). • Systers (Krause & Vingron Bioinformatics 1998, Krause et al. NAR 2002). • PIR (Barker et al. NAR 2000, Wu et al. NAR 2002). • Structural classifications: • SCOP (Murzin et al. JMB 1995). • CATH (Orengo et al. Structure 1997). • FSSP (Holm & Sander NAR 1999). Significant overlap, but no two databases are identical.
Mathematical representations of protein families • Regular expression • Position specific scoring matrices (profiles) • Hidden Markov Models • Probabilistic suffix trees • Sparse Markov transducers
Regular expression G-x(2,3)-[MLIV]-x-P-{K,H}-x(2)-C GAY_LDPAKSC GMF_VEPNTSC GKRFVRPFHNC GKSTVDPFHNC G - a match with amino acid G x – a match with any amino acid x(i,j) – a match with any amino acid at least i times and no more than j times [MLIV] – a match with any amino acid of the four M, L, I or V {K,H} – a match with any amino acid other than K and H
Regular expression - examples ACTININ_1. [EQ]-x(2)-[ATV]-[FY]-x(2)-W-x-N. HOMEOBOX_1. [LIVMFYG]-[ASLVR]-x(2)-[LIVMSTACN]-x- [LIVM]-x(4)-[LIV]-[RKNQESTAIY]- [LIVFSTNKH]-W-[FYVC]-x-[NDQTAH]-x(5)- [RKNAIMW]. HELIX_LOOP_HELIX. [DENSTAP]-[KR]-[LIVMAGSNT]- {FYWCPHKR}-[LIVMT]-[LIVM]-x(2)- [STAV]-[LIVMSTACKR]-x-[VMFYH]- [LIVMTA]-{P}-{P}-[LIVMRKHQ].
Profile (Gribskov et al. PNAS 1987) GAA_TGAAGTC GGT_TTAGGCC GGCAATAGGGC GTTAATAACAC Position specific scoring matrix A 0 0.25 0.25 1 0.5 0 1 0.5 0 0.25 0 C 0 0 0.25 0 0 0 0 0 0.25 0.25 1 G 1 0.5 0 0 0 0.25 0 0.5 0.75 0.25 0 T 0 0.25 0.5 0 0.5 0.75 0 0 0 0.25 0 Add pseudo counts Na(position s)= Ntotal(s)* Σamino acid i p(a/i) p(i/s) Final score: score(a/position s) = Σamino acid i p(i/s) score(i,a)
v1 v2 v3 v4 Hidden Markov Models v1 v2 a22 v3 v4 w2 a11 a12 a23 w1 w3 b11 v1 b14 v2 v3 v4 • Hidden states w and observations v • w(t) – the state at time t • Transition probabilities • P(wj(t+1)/wi(t)) = aij • Emission probabilities • P(vk(t)/wj(t)) = bjk • Transition and emission probabilities are learned from training data
A C G T Hidden Markov Models (cont.) a11 a22 a12 w1 w2 b11 A a21 b14 C T G • A simple intron/exon model for DNA: • Hidden states are w1=exon and w2=intron. • Observations (visible symbols) are A, C, G and T
Hidden Markov Models (cont.) • HMMs used to model protein families are profile HMMs with three different types of hidden states: Match (M), delete (D) and insertion (I) states (Krogh et al. JMB 1996) • The observations (visible symbols) are the amino acids
Hidden Markov Models (cont.) Given a sequence of length T, what is the probability that the sequence was generated by the model? Answer: Sum over all possible paths that can generate this sequence Define the forward variable αj(t) to be the probability to observe the sequence Vt = v(1)v(2)…v(t) and to reach hidden state j at time t Note: P(VT) = Σjαj(T) The HMM forward algorithm: Initialize: αj(1) = P0(j) bjk where vk=v(1) Iteration: for t=1 till T-1 αj(t+1) = Σi αi(t) aijbjk where vk=v(t+1)
Hidden Markov Models (cont.) What is the most probable sequence of hidden states that generated it? The Viterbi algorithm: Define the variable δj(t) to be the probability to observe the sequence Vt = v(1)v(2)…v(t) and to reach hidden state j at time t maximized over all sequences of hidden states of length t-1. Define the pointer variable pathj(t) that contains the pointer to the most probable hidden state at time t-1 from which we can reach state j at time t Initialize: δj(1) = P0(j) bjk where vk=v(1) pathj(1) = 0 Iteration: for t=1 till T-1 δj(t+1) = maxi {δi(t) aij}bjkwhere vk=v(t) pathj(t+1) = arg maxi {δi(t) aij} Output: probability P = maxi {δi(T)} final statewT =arg maxi {δi(T)} and trace back
Probabilistic Suffix Trees (Ron et al. Machine Learning 1996) • Identify short significant patterns (contiguous segments) • Does not require multiple alignment • Induces a probability distribution on the next symbol to appear right after the segment (short term memory) • Variable memory length • More efficient than order L Markov chains • Longer memory length compared to first-order HMMs, and easier to learn
Probabilistic Suffix Trees (cont) • The learning algorithm • Initialize: let tree T consist of a single root node (with empty label) • let S = {σ/ σ in Σ and P(σ) > Pmin } • Building the PST: while S is not empty, pick s in S • Remove s from S • If there exists a symbol σ in Σ such that • P(σ/s) > Tmin • andP(σ/s) > r • P(σ/suf(s)) < 1/r • then add to T the node s and all the nodes on the path to s from the deepest node of T that is a suffix of s • If |s| < L then add the strings {σs / σ in Σ and P(σs) > Pmin } to S • Finalize: smooth tree probabilities { or
Databases of protein domains • Prosite http://www.expasy.ch/prosite/ • Pfam http://www.sanger.ac.uk/Software/Pfam/ • Blocks http://www.blocks.fhcrc.org/ • ProDom http://prodes.toulouse.inra.fr/prodom/doc/prodom.html • Prints http://www.bioinf.man.ac.uk/dbbrowser/PRINTS/ • Domo http://www.infobiogen.fr/services/domo/ • InterPro http://www.ebi.ac.uk/interpro/ • Smart http://smart.embl-heidelberg.de/ • eMotif http://dna.stanford.edu/identify
Motifs and domains • Motif: a simple combination of a few consecutive secondary structure elements with a specific geometric arrangement (e.g., helix-loop-helix). Some, but not all motifs are associated with a specific biological function. • Domain: the fundamental unit of structure folding and evolution. It combines several secondary elements and motifs, not necessarily contiguous, which are packed in a compact globular structure. A domain can fold independently into a stable 3D structure, and it has a specific function. • Domain family: proteins that share a domain (possibly in combination with other domains) • Protein family: proteins that have the same combination of domains
General approaches • Motif based databases • Prosite, Prints, Blocks, eMotif, InterPro • Domain-based databases • Pfam, ProDom, Domo, Smart • Manual/Semi-manual • Prosite • Semi-automatically • Pfam, Smart • Fully automatic • ProDom, Blocks, Domo, eMotif • Use different models (regular expressions, profiles, HMMs) • Based on each other
Prosite • A dictionary of functional and structural motifs and domains • Valuable biological information on each family • Each motif/domain/family is represented as a regular expression, a rule or a profile • Models are generated from (usually published) multiple alignments, manually calibrated to ensure selectivity and sensitivity • Patterns do not always cover complete domains whereas profiles usually span the whole domain • Some families have more than one signature pattern and /or profiles • Pattern length can vary from a few amino acids to several hundreds • As of June 2002 contains 1564 patterns and profiles describing 1144 families or domains 1 2 3 4 5 6 7 8 9 10 11 A 0 0.25 0.25 1 0.5 0 1 0.5 0 0.25 0 C 0 0 0.25 0 0 0 0 0 0.25 0.25 1 G 1 0.5 0 0 0 0.25 0 0.5 0.75 0.25 0 T 0 0.25 0.5 0 0.5 0.75 0 0 0 0.25 0 OR G-x(2,3)-[MLIV]-x-P-{K,H}-x(2)-C
Prosite - example Profile: ID TEST; MATRIX. AC PS50999; MA /DISJOINT: DEFINITION=PROTECT; N1=4; N2=27; MA /NORMALIZATION: MODE=1; FUNCTION=LINEAR; R1=1.1126; R2=0.02183468; TEXT='NScore'; MA /CUT_OFF: LEVEL=0; SCORE=110; N_SCORE=8.5; MODE=1; MA /DEFAULT: M0=-8; D=-20; I=-20; B1=-60; E1=-60; MI=-105; MD=-105; IM=-105; DM=-105; MA /I: B1=0; BI=-105; BD=-105; CC A, B, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, Y, Z; MA /M: SY='R'; M=-12, -9,-30, -9, -6,-24, 10, -8,-33, 16,-24,-13, 0,-19, 0, 36, -7,-13,-23,-20,-17, -6; MA /M: SY='R'; M= -2, -4,-24,-10, -4,-19,-16, -6,-14, 8,-14, -7, 4,-17, 4, 23, -2, -3,-12,-23,-12, -3; MA /M: SY='G'; M= -5, -7,-27, -8, -7,-18, 18,-15,-30, 7,-26,-14, -1,-16, -8, 0, -2,-11,-21,-19,-15, -7; MA /M: SY='G'; M= -9, 4,-27, -2, -9,-24, 24, -6,-32, 0,-27,-17, 16,-20, -6, 12, 0,-12,-27,-25,-21, -9; MA /M: SY='K'; M=-11, -1,-30, -1, 9,-29,-20, -9,-30, 48,-29,-10, 0,-11, 10, 34,-10,-10,-20,-20,-10, 9; MA /M: SY='K'; M= -7, -6,-24, -7, -1,-19, -4,-12,-20, 9,-19,-10, -5,-15, -4, 5, -3, -1,-11,-21,-10, -3; MA /M: SY='V'; M= -1,-30,-12,-31,-30, 0,-31,-30, 32,-21, 11, 11,-29,-29,-29,-21,-11, -1, 48,-29, -9,-30; MA /M: SY='T'; M= -1, -3, 7,-13,-13,-11,-21,-21,-13,-13,-11,-11, -3,-14,-13,-13, 16, 42, -1,-33,-13,-13; MA /M: SY='V'; M= -6,-18,-19,-21,-16, -5,-26,-20, 9,-11, 8, 4,-15,-21,-13, -6, -7, 6, 12,-25, -7,-16; MA /M: SY='V'; M= -4,-30,-17,-33,-29, 1,-33,-29, 35,-24, 17, 14,-27,-27,-26,-23,-15, -4, 40,-26, -6,-29; MA /M: SY='S'; M= -3, 4,-19, 1, 7,-22,-12, -5,-20, 3,-22,-14, 9,-11, 10, 5, 16, 13,-16,-31,-15, 8; MA /M: SY='G'; M= -2, 0,-28, -4,-16,-28, 56,-14,-36,-16,-30,-20, 12,-20,-16,-16, 2,-16,-30,-24,-28,-16; MA /M: SY='L'; M=-10,-29,-21,-33,-23, 14,-31,-21, 23,-28, 36, 20,-27,-28,-21,-21,-25, -9, 15,-17, 3,-23; MA /M: SY='E'; M= -6, 20,-26, 26, 30,-31,-12, -1,-28, 3,-23,-20, 11, -7, 12, -4, 4, -6,-26,-32,-19, 21; MA /M: SY='L'; M= 5,-15,-18,-19, -8, -8,-19,-15, 3,-10, 10, 10,-16,-18, -9,-12, -9, -2, 5,-23, -9, -8; MA /M: SY='F'; M= -6, -7,-21,-11,-12, 21,-18,-10,-12,-10, -9, -9, -4,-21,-17,-11, -5, -5, -9, -9, 11,-13; MA /I: I= -6; MD=-32; MA /M: SY='G'; M= -2, 3,-25, 7, 2,-27, 18,-10,-27, -9,-18,-14, 0,-14, -2,-12, -1,-12,-22,-23,-20, 0; D=-6; MA /I: I= -6; MI=-32; IM=-32; DM=-32; MA /M: SY='I'; M= 0,-21,-20,-25,-20, 4,-25,-20, 17,-22, 16, 12,-19,-22,-18,-21,-15, -7, 14,-19, -3,-20; MA /M: SY='D'; M= -7, 27,-27, 36, 21,-32, -3, -3,-32, 0,-25,-23, 14, -9, 2, -9, 2, -9,-27,-34,-21, 12; MA /M: SY='L'; M= -9,-23,-27,-21,-11, -8,-27,-18, 6,-20, 14, 5,-22, 8, -9,-18,-18, -6, -4,-23,-10,-12; MA /M: SY='K'; M=-11, 2,-27, 3, 10,-22,-20, -4,-24, 23,-21,-11, 0,-12, 10, 16, -4, -2,-19,-19, -3, 9; MA /M: SY='K'; M= 2, -2,-20, -3, 5,-22,-13,-12,-21, 16,-22,-12, 0,-10, 3, 6, 8, 5,-13,-27,-14, 4; MA /M: SY='L'; M= -5,-28,-19,-31,-23, 10,-28,-21, 21,-26, 32, 18,-26,-27,-21,-20,-22, -8, 17,-19, -1,-22; MA /M: SY='A'; M= 31,-14, 0,-22,-15,-17, -2,-22, -8,-15, -6, -8,-13,-17,-15,-21, 2, -3, 1,-24,-19,-15; MA /M: SY='A'; M= 16, -5,-17, -9, -2,-22, -7, -3,-20, 5,-21,-11, -1,-12, 0, 1, 10, 0,-11,-26,-13, -2; MA /M: SY='E'; M= 3, 4,-23, 5, 13,-23,-16, -9,-13, -3,-12, -7, -2, -9, 5,-10, 1, -2,-12,-27,-15, 9; MA /I: E1=0; IE=-105; DE=-105; Pattern: K-x-V-[TC]-x-[IVL]-x(2)-[LMF] Courtesy of Nicolas Hulo
Prints • A database of gene- and domain-family fingerprints in the form of collections of short motifs aligned without gaps • Fingerprints comprise several motifs from different parts of a multiple alignment • Discrimination power increases with the number of motifs in a fingerprint • Start from a seed alignment of a few sequences. Search the database and refine the fingerprint based on the set of sequences that match the fingerprint completely (add sequences and/or add/remove motifs), until convergence • Partial match – possibly subfamily members • As of July 2002, contains ~10,500 motifs grouped into 1750 families
Prints - a fingerprinting overview PRINTS annotation N C Courtesy of Terri Attwood
Prints - example SUMMARY INFORMATION 37 codes involving 8 elements 0 codes involving 7 elements 0 codes involving 6 elements 0 codes involving 5 elements 0 codes involving 4 elements 1 codes involving 3 elements 0 codes involving 2 elements COMPOSITE FINGERPRINT INDEX 8| 37 37 37 37 37 37 37 37 7| 0 0 0 0 0 0 0 0 6| 0 0 0 0 0 0 0 0 5| 0 0 0 0 0 0 0 0 4| 0 0 0 0 0 0 0 0 3| 1 0 0 0 1 1 0 0 2| 0 0 0 0 0 0 0 0 -+----------------------------------------- | 1 2 3 4 5 6 7 8 True positives.. PRIO_COLGU PRIO_MACFA PRIO_CEREL PRIO_ODOHE PRIO_GORGO PRIO_PANTR PRIO_HUMAN O46648 PRIO_SHEEP PRIO_CALJA PRIO_BOVIN PRP2_BOVIN PRIO_ATEPA PRIO_SAISC PRIO_PREFR PRIO_PONPY O75942 PRIO_CAPHI PRIO_CEBAP PRIO_CAMDR PRIO_FELCA PRP1_TRAST PRIO_RABIT PRP2_TRAST PRIO_PIG PRIO_CANFA PRIO_CRIGR PRIO_CRIMI Q15216 PRIO_RAT PRIO_CERAE PRIO_MUSPF PRIO_MUSVI PRIO_MESAU PRIO_MOUSE O46593 PRIO_TRIVU Subfamily: Codes involving 3 elements Subfamily True positives.. PRIO_CHICK Courtesy of Terri Attwood
Prints - website Courtesy of Terri Attwood
ProDom • A database of automatically generated protein domains • Old procedure: • Identify high-scoring segment pairs (HSPs) using BLAST for all-versus-all comparison of SwissProt • Construct homologous segment sets by transitive closure of overlapping HSPs • Break into domains • New procedure: • Construct homologous segment sets by PSI-BLAST recursive search • Generate a multiple alignment and a consensus sequence for each family • May break real domains into smaller domains (more than 305,000 domain families as of January 2002)
ith iteration DB query internal repeat detection yes no query PSI-BLAST no match matches repeat matches DB changes remove newly found domains split modified sequences sort by size (i+1)th iteration DB query Courtesy of Daniel Kahn
ProDom - website • Domain analysis • Which proteins contain a given domain? • Which proteins share a homologous domain with a given protein? Domain motif Links to other representations of the family Summary alignment Parameters to define sub-family levels Courtesy of Daniel Kahn
ProDom - website • Synthetic views of alignments and trees • Homology search with graphical output Courtesy of Daniel Kahn
Links to SWISS-PROT, TREMBL, PROSITE & PDB • Links to 3-D modelling with SWISS-MODEL Courtesy of Daniel Kahn
Blocks • A collection of blocks (short ungapped alignments) • Each represent a conserved region of a protein family (not necessarily associated with a known function) • Blocks are generated from multiple alignments of groups of related proteins. • Groups are derived from protein families in Prosite, Smart, Pfam, ProDom • Used only the SWISSPROT entries to define the blocks (to avoid false positive sequences from TrEMBL) • Complementary to PRINTS • As of August 2001 contains 8656 blocks representing 2101 groups
Blocks (cont) GAYKLDPAKSC GMFMVEPNTSC GKRFVRPFHNC GKSTVDPFHNC • Detection: • Identify short motifs using the algorithm by Smith 1990 • Short patterns are then extended in both directions until the similarity score drops. Maximal length of a block is 60 • Assembly • Create an ordered set of non overlapping blocks (“path”) that occur in a critical number of sequences • Scoring • Score individual blocks based on length, similarity, #sequences, etc • Score a path based on scores of the constituent blocks and #sequences DKGAYKLDPAKSCNL HSGMFMVEPNTSCHF GLGKRFVRPFHNCWM MMGKSTVDPFHNCHD DKGAYKLDPAKSCNL HSGMFMVEPNTSCHF GLGKRFVRPFHNCWM MMGKSTVDPFHNCHD KHAYKLDAKSNLL VHMFMVDNTSHFL LHKRFVDFHSWMM KKVLDEAKSCN KLILEENTSCH SLFLREFHNCW AAVLDEFHNCH SKLLEEFSECH
GAY_LDPAKSC GMF_VEPNTSC GKRFVRPFHNC GKSTVDPFHNC Database build search refine Pfam • A database of Hidden Markov Models for protein domains/families generated semi-automatically • Start from a seed multiple alignment (published or from other databases, such as Prosite and ProDom). • The alignment usually represent complete domains (confirmed with structural data when available). • After manual inspection a profile HMM is built from the alignment • The HMM is used to search the database. If a true member is missed, then it is added to the seed alignment and the process is repeated. • A full alignment is created for all members of the family. If it is not correct the process is repeated with another seed alignment or another alignment method • As of May 2002, contains 3849 families, domains, repeats and motifs (in addition Pfam B derived from ProDom)
Domo • Combines compositional and local similarity search followed by multiple sequence alignments • Start by detecting global similarities from the comparison of amino acid and dipeptide composition of each protein. • Group similar proteins into clusters • Pick one representative from each cluster and compile into a suffix tree • Compare the suffix tree with itself local similarities • Cluster local similarities and align (without gaps) • Analyze alignments to detect domain boundaries and split • Create multiple alignments • Iterative family extension using automatically generated regular expressions • Contains 8877 entries
InterPro • Combined resource of proteins domain and motifs from ProSite, ProDom, Prints, Smart and Pfam
Databases of protein clusters • Cogs http://www.ncbi.nlm.nih.gov/COG/ • Systers http://systers.molgen.mpg.de/ • PIR http://wwwnbrf.georgetown.edu/pirwww/pirhome.shtml • ProtoMap http://protomap.cornell.edu/ What are the differences?