Download

1 / 1
10 likes | 116 Views
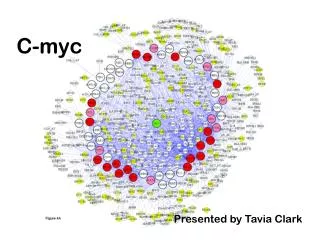
c-Myc Regulated Functional Gene and Protein Networks Involved in Tumourigenesis. Sam Robson. 2: Pancreatic Cancer and Diabetes

E N D
c-Myc Regulated Functional Gene and Protein Networks Involved in Tumourigenesis Sam Robson 2: Pancreatic Cancer and Diabetes The pancreas is responsible for regulating blood glucose levels. It contains hundreds of colonies of cells within the exocrine tissue, known as islets. One of the cell types contained within the islets are β-cells, the main source of insulin within the body. Excessive proliferation of β-cells results in insulinoma, while excessive apoptotic activity has been linked to some forms of diabetes (due to the subsequent loss of insulin, leading to a drop in glucose metabolism). 3: Transgenic Model By the time cancers are detected, they are usually very advanced and have acquired many additional genetic lesions. This makes it difficult to identify the initial pathways involved in tumourigenesis, making human in vivo study difficult. Mouse strains have been genetically modified to allow direct control of the activation of the c-Myc transcription factor within the pancreatic islets. This allows the analysis of the genetic changes that occur as a direct result of c-Myc activation. To do this, c-Myc is fused at the carboxy-terminal domain to the hormone binding domain of a mutant mouse oestrogen receptor to form a c-MYC-ERTAM transgene. This transgene encodes a chimeric protein c-MYC-ERTAM that can be activated by the specific ligand 4-hydroxytamoxifen (4-OHT), which is administered through daily intraperitoneal injections. 1: C-Myc Deregulation of the c-Myc (Carcinoma Myelocytomatosis) proto-oncogene is seen in a large number of human cancers, resulting in aggressive, poorly differentiated tumours. The c-Myc protein is a transcription factor, and is known to be involved largely with cell cycle progression (G1 to S phase) and the inhibition of terminal differentiation. Paradoxically, the transcription factor also appears to sensitize the cell to apoptotic activity. c-Myc works as part of a heterodimeric complex with the protein Max, which binds itself to the carboxy-terminal basic-helix-loop-helix-zipper (bHLHZ) domain of the c-Myc protein. This Myc-Max heterodimer is able to bind specific DNA sequences, such as the E-box sequence ‘CACGTG’. We also find two highly conserved elements in the N-terminal domain; the ‘Myc boxes’ MBI and MBII. These are required for transactivation of various target genes. Activation of the c-MYC-ERTAM protein in the islets results predominately in apoptosis rather than proliferation, leading to islet involution and onset of diabetes within 9 days. This indicates that the β-cells are only mildly buffered against cell death in vivo. In order to study the oncogenic potential of c-Myc in islet carcinogenesis, Myc-induced apoptosis was blocked by constitutive over-expression of the anti-apoptotic protein Bcl-XL (Beta-cell lymphoma X - large). This was achieved by cross-breeding the c-Myc-ERTAM mice with transgenic mice in which expression of Bcl-XL is placed under the control of the rat insulin promoter (RIP7). This produces double transgenic Ins-c-Myc-ERTAM/RIP-Bcl-XL, otherwise known as RM mice. Activation of c-MYC-ERTAM in double transgenic RM mice results in rapid, synchronous entry of nearly all β-cells into the cell-cycle with no discernable c-Myc induced apoptosis. The islets continue to increase in size so long as 4-OHT is continuously administered. Thus c-MYC-ERTAM activation results in grossly hyperplastic islets . Carcinogenesis involves accumulation of several somatic lesions, leading to unmediated growth of cells, loss of differentiation, invasion and angiogenesis. By controlling the expression of c-Myc and preventing cell death, we can study the role that it plays in these processes. Microarrays are high density oligonucleotide chips that are able to measure the expression levels of every gene within the genome by measuring RNA levels. Affymetrix Genechips are commercially available microarrays with very high reproducability. 4: Microarrays 5: Microarray Data Analysis Microarray experiments create a huge amount of data. At least three replicate chips must be run for each time point to allow statistical analysis. Four time points were used in this experiment; Time 0 (with no 4-OHT administered), 1 day and 14 days of 4-OHT administration, and a tumour reversal time point, with 4-OHT administered for 14 days, then stopped for 7 days. The Microarray data was analysed using Genespring, a very powerful piece of software from Agilent. The first step of data analysis is to perform quality control to remove outlying samples. Several methods can be used, such as hierarchical clustering algorithms and principle component analysis. The data must also be normalised to allow comparisons between genes on different chips. Normalization shifts the data to have mean 1. The next step of analysis is to reduce the dimensionality of the data by finding ‘important’ genes. These are genes that are differentially expressed across the time course, and may provide an insight into the pathways involved at various stages of tumourigenesis. RNA is extracted from pancreatic tissue at various timepoints from the start of 4-OHT administration. The RNA is labelled with biotin and washed onto the chip (MOE430 Plus2 Mouse chip). RNA strands bind to the oligonucleotide strands on the chip. Oligonucleotides matching to specific genes are organised together to form probes. A fluorescent image of the chip is taken, where the intensity of each of the probes relates to the concentration of RNA in the cell. One of the key methods of analysis is to map the expression profiles onto pathways available from the Kyoto Encyclopaedia of Genes and Genomes(KEGG). Shown here are the expression levels of genes involved in the cell cycle after 1 day of 4-OHT administration (red = upregulation; blue = downregulation; yellow = no change). Pathways are available for many of the most important processes of the body. By analysing changes in gene expression levels at various timepoints, we get an idea of the effect that c-Myc has on these pathways. One key feature being focused on is the DNA damage pathway, and how this relates to the apoptotic activity of c-Myc. The exact mechanisms of how c-Myc leads to apoptosis is currently unknown, and it is hoped that this will shed some light on the problem. 6: Conclusions and Future Research Whilst many conclusions can be made from this data, it is uncertain how relevant they will be. Much of the data is variable, suggesting problems with the collected RNA. Also, many genes show unexpected expression profiles. Whilst this may show novel gene targets of c-Myc, it is more likely that this is due to problems with the RNA (be it RNA degradation, masking of important data by exocrine tissue or the non-comparability between the time points). The group is currently working to solve this problem by using Laser Capture Microscopy to isolate islet cells for microarray analysis. Current problems exist due to RNA degredation, which we hope to solve in the near future. Once this problem has been overcome, a similar microarray experiment will be run focusing on early timepoints. This will allow the analysis of direct effects of c-Myc activation in the islet tissue, allowing the distinction of direct c-Myc targets from indirect targets. However it has been suggested that as many as 10% of the genes in the genome are under the direct transcriptional control of c-Myc (close to 4,500 genes), which may make this process difficult. Once information on c-Myc targets, along with downstream effects, has been discerned, gene network pathways can be set up showing the effects of c-Myc activation on tumourigenesis. Various mathematical techniques are currently being tested for the validation of these pathways, most notably the Bayesian approaches being optimised by the IPCR group. Once these networks have been fully understood, it is hoped that they may provide possibilities for therepeutic research in the future. We see much variation in the data, which suggests that the RNA quality is not as good as it could be. The pancreas contains many RNAses (enzymes that degrade RNA), that begin to act instantly upon making incisions into the tissue. Several methods have been used to prevent this degradation, including snap freezing the tissue in liquid nitrogen instantly after extraction, fixing the tissue using formalyn, and using the RNAlater stabilising reagent from the Qiagen company. Currently, work is under way to improve these methods, such as perfusing formalyn into the tissue through the blood system before extraction. A further problem with the data is a result of using the entire pancreatic tissue when all that we are interested in is the islet tissue. The islets make up only a small part of the total tissue mass and it is probable that exocrine pancreatic tissue will mask the RNA levels of the islets. Another problem comes from the fact that the islet to exocrine ratio is not the same at the various time points. Thus comparisons between expression levels across the time points may not be useful. Further studies should be performed using only RNA from the islet cells. Acknowledgements Special thanks to Mike Khan and Stella Pelengaris for giving me this opportunity to work with their group, and also to David Epstein for keeping me routed in the maths department. Thanks also to Helen Bird, Lesley and Sue Davis for their help with the Microarray work. Lastly, thanks to Linda Cheung, Vicky Ifandi, Sevi, Göran and Sylvie Abounafor all of their help in the lab work.