Download
1 / 70
780 likes | 1.3k Views
Hemostasia y Trombosis. Dra. Fabiola Navarro C. HEMOSTASIA "Todos aquellos mecanismos que tienden a evitar la pérdida de sangre por extravasación implicando también los mecanismos de disolución del trombo formado una vez cumplida su misión” Vasos sanguíneos y componentes de su pared

E N D
Hemostasia y Trombosis Dra. Fabiola Navarro C.
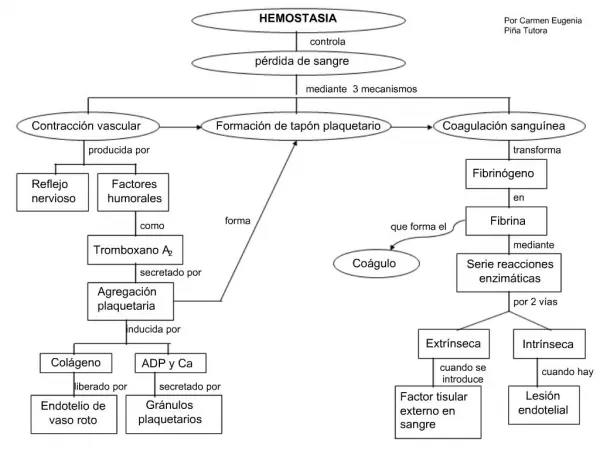
HEMOSTASIA • "Todos aquellos mecanismos que tienden a evitar la pérdida de sangre por extravasación implicando también los mecanismos de disolución del trombo formado una vez cumplida su misión” • Vasos sanguíneos y componentes de su pared • Plaquetas circulantes • Factores de coagulación solubles en el plasma • Micropartículas "Los elementos participantes deben estar cualitativamente y cuantitativamente normales"
Secuencia del proceso hemostático Ruptura de un vaso Contacto del subendotelio con plaquetas Adhesión plaquetaria Activación plaquetaria---(tromboxano A2, secreción de gránulos) Sello plaquetario primario Activación sistema de coagulación En plaquetas expresión de receptores de factores de coagulación Reacción y localización de formación del coágulo (hemostasia secundaria) Reabsorción de la fibrina (fibrinolisis) Remoción de restos celulares -reparación tisular- Reendotelización del vaso sanguíneo
Coagulación • 1964 : “ la coagulación se logra a través de una serie de reacciones proteolíticas que se llevan a cabo de manera secuencial o en cascada ”. • Se plantearon dos sistemas para interpretación en el laboratorio : • Vía intrínseca y extrínseca.
Coagulación • Células • Proteinas coagulantes • Receptores celulares • Proteasas • Cofactores ( FV y FVIII )
Factores de coagulación - Nomenclatura Nomenclatura internacional y función de los factores de la coagulación Factor Sinónimo Función I Fibrinógeno Estructural II Protrombina Serina proteasa III Tromboplastina tisular Cofactor/iniciador IV Calcio V Factor Labil, proacelerina Cofactor VI No asignado VII Proconvertina Serinoproteasa VIII Factor antihemofílico IX FactorCristmas-antihemofílico B Serinoproteasa X Factor Stuart Serinoproteasa XI Factor antihemofílico C Serinoproteasa XII Factor de Hageman Serinoproteasa PK Precalicreina Serinoproteasa HMWK Cininógeno APM Cofactor XIII Factor estabilizador de la fibrina Transglutaminasa
Coagulación • La mayoría de los procesos de activación se dan sobre la superficie celular. • Presencia de fosfolípidos de membrana • Expresión de receptores de proteinas • Anclaje de factores de coagulación ; superficie aniónica de ensamble. “ teoría celular de la hemostasia ” 2003.
Modelo celular de la hemostasia • Iniciación • Amplificación • Propagación
Modelo celular & Iniciación • Factor tisular : - proteina de membrana - células extravasculares ( fibroblastos) - célula endotelial y monocitos( inflamación -expresión frente a daño endotelial
Modelo celular & Iniciación • FT + FVII • FT/FVIIa • Activación FIX---------FIXa FX------FXa • FXa + FVa ( protrombinasa) • Protrombina------------ trombina ( escasa) • Activación FV- FVIII y plaquetas • Fase de amplificación
Modelo celular& Amplificación • Contacto entre plaquetas y componentes plasmáticos ( ruptura de vaso) • Adhesión a colágeno / FvW • Activación plaquetaria y localización en zonas de exposición de factor tisular. • Trombina ----------amplificación
Modelo celular& Amplificación • Aumenta adhesión plaquetaria • Activación plaquetaria y agregación irreversible • Activación de FV-FVIII-FIX por trombina. • Trombina unida a receptores plaquetarios provoca ensamble de complejos procoagulantes “tenasa” IXa/VIIIa,y “protrombinasa”Xa/VA sobre la superficie plaquetaria. • Propagación
Modelo celular& Propagación • Ensamble complejo FIXa/FVIII (superficie plaquetaria) • Activación FXa • FXa/FVa • “Estallido de trombina”---------Fibrina soluble • FXIa----IXa+FVIII • FXa • Fibrina s + FXIIIa + calcio-----fibrina insoluble.
COAGULOPATÍAS • Enfermedades del sistema hemostático. Historia Clínica: Pacientes que refieren historia de sangrados anormales , no relacionados a traumatismos, o sangrados excesivos frente a trauma menor. • Puede haber antecedentes familiares • Buscar historia desde el nacimiento • No olvidar que aún sin historia previa pueden presentarse sangrados anormales descubiertos tardíamente y aún con pruebas de coagulación normal • Ingesta de fármacos- alcohol • Enfermedades concomitantes
Examen físico: - Palidez - Petequias - Lesiones purpúricas - Equímosis, hematomas - Epistaxis, gingivorragias - Hemorragias de mucosas - Hemartrosis
Laboratorio Laboratorio de Tamizaje - Extendido de sangre periférica---- recuento de plaquetas - Tiempo de sangría - Tiempo de protrombina (PT) - Tiempo de tromboplastina parcial activada (PTTA) - Tiempo de Trombina - Solubilidad del coágulo - Inhibidores adquiridos de coagulación ( anticoagulante lúpico) Laboratorio Específico - Estudio de Agregación y secreción plaquetaria - Estudio de adhesividad plaquetaria - Medición de Factores de Coagulación
COAGULOPATÍAS Adquiridas Hereditarias
Coagulopatías Hereditarias de la Coagulación - Hemofilias - Enfermedad de Von Willebrand - Déficit de otros factores
Hemofilias • Enfermedad hereditaria ligada al sexo, transmitida por la mujer y solo manifiesta en el hombre. • 40 % de los casos no tiene antecedentes familiares conocidos. • Tendencia hemorrágica a temprana edad y con sangrados incoercibles. • Enfermedad crónica causante de un amplio espectro de complicaciones y secuelas graves. Clasificación: - Hemofilia A Factor VIII(15-20/10.000 ) - Hemofilia B Factor IX(1,5 / 100.000) Leves Moderadas Severas Diagnóstico : PT prolongado o N Ptta prolongado o N Cuantificación de Factores
Hemofilias & Clasificación SEVERA 0 - 1 % ACTIVIDAD MODERADA 1 - 4 % ACTIVIDAD LEVE 5 - 24 % ACTIVIDAD SUBNORMAL 25 -49 % ACTIVIDAD NORMAL mayor 50 % ACTIVIDAD
Tratamiento • Educación • Apoyo psicosocial • Apoyo fisiátrico y kinésico • Aporte de Factor deficiente ( liofilizados) • Desmopresina • Crioprecipitados • Plasma Fresco Congelado (PFC) • Ejercicio permanente y sistemático supervigilado, fortalecimiento muscular Articulaciones protegidas.
Contraindicaciones • Punción intramuscular • Punciones, procedimientos invasivos sin terapia de reemplazo IMPORTANTE “El paciente con hemofilia debe ser manejado como cualquier paciente , APORTÁNDOLE previamente la terapia de reemplazo que corresponda. "
Enfermedad de von Willebrand • El factor FvW es una glicoproteina de alto peso molecular sintetizado y almacenado en megacariocitos y células endoteliales. • La estructura del FvW polipéptido que en cada subunidad contiene sitios de unión para colágeno y para las glicoproteinas Ib y IIb/IIIa. • Cuando hay daño en un vaso , éste expone el subendotelio y se une el FvW, esto conlleva a un cambio en el FvW que expone los sitios de unión para que la glicoproteina Ib de las plaquetas se una al FvW y se lleve a cabo el mecanismo de adhesión plaquetaria. • El FvW se adhiere a colágena de la pared vascular y activa el sitio de unión dela GP IIb/IIIa ocurriendo la agregación plaquetaria, por medio del FvW, fibrinógeno y otras proteinas. • También el FvW funciona como acarreador del F VIII permitiendo la estabilidad de este factor en la circulación.
Enfermedad de Von Willebrand • Enfermedad hemorrágica hereditaria, autosómica dominante. • Heterocigota ( frecuentes). Homocigota ( raras). • Prevalencia: 0,8% • Patogenia: Disminución cuantitativa y/o funcional del factor von Willebrand plasmático. • Clasificación • E vWillebrand Tipo 1 • E vWillebrand Tipo 2 • 2A • 2B • 2M • 2N • E v Willabrand Tipo 3
Cuadro Clínico • Hombres/Mujeres • Epistaxis, equímosis, alveolorragia, gingivorragia. • Hemorragia prolongada en heridas menores. • Hemorragia intra o postcirugías. • Hemorragias postparto. • Menorragias. • Hemorragias gastrointestinales. • Curso cíclico.
Laboratorio • Tiempo de sangría prolongado o normal. • Medición de FvW. • Medir actividad de FvW ( cofactor ristocetina). • Identificación de estructura multimérica.
Tratamiento • Educación • No Aspirina- AINES • Antifibrinolíticos • DDAVP : 0,3 mcg/ k • Crioprecipitados • Profilaxis ante eventos quirúrgicos o procedimientos. • F vW
TRANSTORNOS ADQUIRIDOS DE LA COAGULACIÓN • Déficit de Vitamina K. • Enfermedad del RN • Abuso de antibióticos de amplio espectro. • Sindrome de Malabsorción. • Anticoagulantes orales. • Abuso de laxantes a base de aceite. • Obstrucción de vía biliar.
Enfermedades Hepáticas : • - Cirrosis hepática OH • - Hepatitis crónica • Consumo de Factores de Coagulación (Ej. CID) • Pérdida de Factores • - Sd. Nefrótico • - Amiloidosis • Inhibidores patológicos de la coagulación.
CID • La Coagulación Intravascular Diseminada (CID) constituye unsíndrome trombohemorrágico adquirido con daño orgánico deintensidad variable y que presenta evidencia de laboratorio dehiperactivación de la coagulación, consumo de inhibidores y actividadfibrinolítica, además de presencia de anemia hemolíticamicroangiopática. • Las situaciones clínicas que provocan daño endotelial y tisular son las que con mayor frecuencia se asocian a CID. • La patología responsable por exelencia del daño endotelial es la sepsis de cualquier origen.
Causas de CID * GINECOLÓGICAS : - Embolía de líquido amniótico. - Desprendimiento de placenta. - Eclampsia. - Huevo muerto retenido. * CANCER : -Leucemia promielocítica. -Metástasis * HEMÓLISIS INTRAVASCULAR: - Sd. de transfusión incompatible. * INSUFICIENCIA HEPÁTICA AGUDA * SEPTICEMIA
CID & Manifestaciones Clínicas • Síntomas variables e inespecíficos : fiebre, hipotensión, acrocianosis, gangrena, hemorragias mucocutáneas y en sitio de heridas quirúrgicas y de venopunción, mal estado general y datos de daño orgánico múltiple. • El cuadro clínico de CID fulminante, se caracateriza por trombosis y hemorragia a la vez y se observa comunmente en sepsis por Gram (-) ( Sd. de Waterhouse Friderichsen).
CID & Laboratorio • Recuento de plaquetas menor a 50.000. • Presencia de esquistocitos en frotis hemograma. • PT prolongado. • TT prolongado. • Fibrinógeno menor a 200 mg/dl. • PDF elevados • Dímero D elevados
CID & Tratamiento • Soporte hemodinámico. • Soporte transfusional : aporte de GR, PFC, plaquetas. • Terapia antitrombótica? • Antitrombina III, Concentrados de proteina C activada.
SINDROMES PURPÚRICOS Definición: Manifestaciones hemorrágicas en la piel y en las membranas mucosas que se manifiestan como petequias y equímosis. Puede haber episodios de sangrados gastrointestinal, menorragias y/o hematuria.
Clasificación Púrpuras ( Recuento de plaquetas) Trombocitopénicos No Trombocitopénicos • Central • Periférico
Púrpuras Trombocitopénicos • Inmunológico Idiopático (PTI). • Enfermedades Autoinmunes (LES). • SIDA. • Drogas. • Post-transfusional. • Hiperesplenismo. • Producción medular dismimuida.
Púrpuras No Trombocitopénicos • Simple. • Mecánico. • Ortostástico. • Senil. • Alérgicos. • Vasculitis. • Transtornos funcionales plaquetarios. • Adquiridos : Uremia ,drogas (AINES), • paraproteinas. • Congénitos : Trombastenia de Glazmann, E • Enfermedad de Bernard-Soulier.
Púrpura Trombopénico Inmunológico (PTI) • Enfermedad caracterizada por la presencia de trombocitopenia mediada por anticuerpos antiplaquetarios. La médula ósea mantiene producción normal o aumentada de megacariocitos.