Genetic Population Subdivision & Mixing Effects
320 likes | 346 Views
Explore population genetics concepts like Wahlund effect, F-coefficients, and gene flow in mixed populations. Understand the impact of genetic drift, mutations, and local adaptations on allele frequencies. Learn about FIS, FST, and FIT coefficients and their calculation principles. Discover how population structure influences genetic variance and gene flow. Gain insights into genetic structuring in different species and its implications on FST values. Delve into the genetic structuring of fish populations compared to humans.

Genetic Population Subdivision & Mixing Effects
E N D
Presentation Transcript
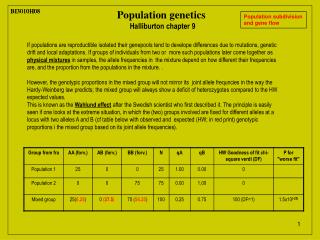
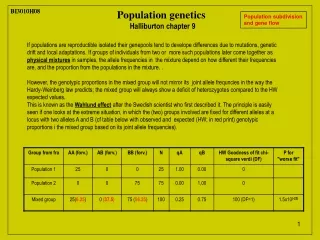
BI3010H08 BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow If populations are reproductible isolated their genepools tend to develope differences due to mutations, genetic drift and local adaptations. If groups of individuals from two or more such populations later come together as physical mixtures in samples, the allele frequencies in the mixture depend on how different their frequencies are, and the proportion from the populations in the mixture. . However, the genotypic proportions in the mixed group will not mirror its joint allele frequncies in the way the Hardy-Weinberg law predicts; the mixed group will always show a deficit of heterozygotes compared to the HW expected values. This is known as the Wahlund effect after the Swedish scientist who first described it. The principle is easily seen if one looks at the extreme situation, in which the (two) groups involved are fixed for different alleles at a locus with two alleles A and B (cf table below with observed and expected (HW; in red print) genotypic proportions i the mixed group based on its joint allele frequencies).
BI3010H08 BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow Population structure and F coefficients (Wright 1951) Consider a large population subdivided in many smaller populations, and a single locus with two alleles A1 and A2. (See nomenclature on pages 318 and 319). The mean deviation in heterozygosity among subpopulations is: FIS = (HS – HI) / HS where HI is the mean of the observed heterozygosities in subpopulations, and HS is the mean of the HW-expected heterozygosities. The subscript IS means deviation among individuals relative to subpulation. Within subpoplations the evolutionary forces are in action, and heterozygosity can be higher or lower than HW-expected values (i.e. be positive or negative, from +1 to -1). The deviation in heterozygocity due to population subdividion is: FST = (HT – HS) / HT where HT indicates the expected heterozygocity in the total population, and HS indicates the mean of expected heterozygocity in the subpopulations. The subscript ST means deviation in heterozygocity among subpopulations relative to the total population. Subdivision always means a apparent underreprentation of heterozygotes (Wahlund effect). Therefore, FST is always either zero or positive, and can vary from 0 to one. The deviation in heterozygosity in the total population is: FIT = (HT – HI) / HT where HT indicates the expected heterozygosity based on the total allele frequencies, and HI indicates the observed heterozygosity in the total population (which is the mean of observed heterozygosities in the subpopulations. Subscript IT indicates the deviation among individuals relative to the total population ( -1<FIT<1, and depends on the values of FISog FST). See Box 9.2 for practical calculation of F-coefficients.
BI3010H08 BI3010H08 Population subdivision and gene flow Population genetics Halliburton chapter 9 The three F-coefficients are not additive, but tied together by the following relation: (1 - FIT) = (1 – FIS)*(1 – FST) (Box 9.3 page 321). F-coeffiicients for multiple alleles are calculated after the same principles as for two alleles. The calculations can be work- consuming, and are usually made by computers. F-coefficients for multiple loci are calculated as mean values over loci. Different interpretations of FST: Above,FSTwas defined in terms of expected heterozygosities within and between subpopulations. The coefficient can also be seen as the proportion of the total heterozygosity in the total population that is due to allele frequency differences between subpopulations. Wright (1951) and Weir & Cockerham (1984) have different approaches. A useful understanding of FST is the proportion of the total genetic variance that is due to differences between subpopulations. (The rest of the variance is due to differences between individuals if we consider a simple stucture with only two levels; the total population and the subpopululations). Modern software make it practically possible to calculate the "contributions" from many more levels, for example area, region, sex etc). The calculation is called Manova and is a parallell to Anova. Experience has shown that the size of FST between selfrecruiting populations is strongly affected by the species' general biology (population size, geographical distribution range, homing tendency etc). With other words; what the possibilities are for a gene flow between them. In fish for example, limnic species show a stronger structuring than anadromous, which again are more structured than marine fishes. It may be mentioned as a curiocity that typical FST for salmon polulation are approximately the same as for the main races in humans (mongolid, negroid, caucasions, pygmees and aborigins), i.e. FST around 0.10). A very valuable property of FST is that it is a relative measure (range 0-1), meaning that FST are comparable even though they may be based on different types of genetic markers (e.g. Isozymes and DNA markers) with different mutation rates.
BI3010H08 BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow The statistical significans of calculated FST values: Many PC-programs estimate confidence intervals for FST, enabling decisions on whether it is different fom zero. Other programs calculates the factical P-value for this. These tests are often based on certain assumptions (like equally sized subpopulations). The test with most "power" in testing the statistical "certainty" of an observed heterogeneity between populations (or samples from populations) is still, however, based on the actual genotypic or allelic number in the samples. This test is the chi-square homogeneity test (or chi-squared RxC contingency table test). Testing for allelic proportions has more power than tests of genotype proportions if we have HW-proportions in subsamples (see examples next page). The null hypothesis under test is that the samples could be taken from the same populations, or from populations with the same genetic characteristics at the locus under test, and that the differences could be caused by random effects during sampling. On the next page we first test for differences in genotype proportions, and then for differences in allelic proportions.
BI3010H08 BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow Principle for calculating expected genotype- and allele numbers under the null hypothesis. E.g. For genotype AA in sample 1: (108 / 380)x80=22.7 E.g. For allele A in sample 1: (382 / 760) x 160 = 80.4 Under the null hypothesis it is the numbers under "Total" that is our best estimate of the true distribution. Chi-squared test of homogeneity of genotypic proportion.( RxC chi-squared contingency table test). Chi-squared total for 9 cells = 59.574. DF = (R-1) x(C-1) = 2x2 = 4. P << 0.001 Chi-squared test (RxC) of homogeneity of allele proportions Chi-squared is calculated as always, i.e. as (O-E)2 / E for each cell, and then summed. Degrees of freedom in RxC tests are: (R-1) x (C-1) R = no. of "Rows" C = no. of "Columns" Chi-squared total for 6 cells = 47.735 DF = (R-1) x(C-1) = 2x1 = 2. P << 0.001
BI3010H08 BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow Gene flow is defined as an immigration of individuals (genes) from one population to another, with subsequent reproduction. Its symbol, m, denotes the proportion of reprodusing individuals that are immigrants. If the allele frequencies of immigrants and locals are different, the gene flow (immigration) will lead to a reduction of the differences at all polymorphic loci, to a degree that depends on the proportion of immigrants (m). "Continent-Island model" This is the simplest model of gene flow. It occurs as a one-way gene flow from a large Mainland population to s small Island population. The Mainland population is thought to have no changes in its allele frequencies, while those in the Island will change over time due to the immigration. Generally, the model covers any immigration to a local population (recipient) from a non-local (donor). The effect of the immigration on local allele frequency can be described as: pt+1 = (m)( pm ) + (1-m )(pt ), and p = m( pm – pt ) "Island model" This model is figured as a system of islands which exchange individuals; i.e. the gene flow is resiprocal and random. The gene flow to each island population thus depend on the proportion of individuals from the various other islands. The islands have different allele frequencies, the allele frequency of the immigrants will be the weighted mean of those of the donor islands involved. The development over time is described by the recursion pt+1 = (m)( psnitt ) + (1-m )(pt) "Stepping stone" models These can be one- or two-dimensional (Fig. 9.3 c,d). Populations exchange genes primarily with neighbouring populations each generation, hence neighbouring populations will be genetically more similar than distant ones. A variant of this model is the "Isolation by distance model", in which the individuals can be spread over large geographicc distances, so that the probability that individuals meet for mating is reduced by increasing distances. (e.g. as in polar bears Fig. 9.6).
BI3010H08 BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow Gene flow and differentiation Random genetic drift makes populations diverge genetically, while gene flow counteracts this. How much gene flow is needed to counteract a certain genetic drift? The answer depends on which model for geneflow is chosen, and the actual strength of the various evolutionary forces. However, exclusing other forces the realtion between genetic drift (i.e. population size) and gene flow is surprisingly simple. Measured i terms of FST the equilibrium situation is described by this formula: which says that in the equilibrium situation the differentiation depends only on the relative strength of the product Mn, which is the absolute number of immigrants per generation. This may look non-intuitive, but follows from the fact that in small populations with large genetic drift, a larger proportion of immigraats is needed to balance the genetic drift effect than in larger populations with less genetic drift. The approach to the equilibrium is assymptotic and will take a large number of generation to reach (approx. 4N generations where N is the effective population size). For many natural populations an equilibrium is never reached, but neverthess formula 9.15 is frequently (too often?) employed for estimating immigration rates from an observed FST value. However, underway towards equlibrium, observed FST is smaller than equilibrium value and will thus of course over-estimate the gene flow. The graphs on the next pages show the differentiation of allele frequencies as a function of Nm, and the relation between H and Nm.
BI3010H08 BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow
BI3010H08 BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow
BI3010H08 BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow
BI3010H08 BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow By DNA sequencing (e.g. of haploid mtDNA) the genealogy of the gene can be disclosed, that is; the history of the mutations from a common basic form. This reveals unique information for use in studies of the distribution and spreading history of species. The basis for this approach is formulated in coalescence theory.
BI3010H08 BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow GENETIC DIFFERENTIAION The table below shows the genotype distribution at a polymorphic locus with two alleles A and B from two populations. The genotype proportions are chosen so that each population is in perfect HW equilibrium. The bottom line shows the genotypic proportions and allele frequencies in a 50:50 mix of the two. Note thatthe physical mixture (Total) shows a deficit of heterozygotes compared to the expected valued based on the joint allele frequencies. Recall that this is due to the Wahlund effect. In the following we shall use the tabulated values to calculate various measures for genetic differentiation. The first one is a relative measure (Sewall Wright's FST, which has several analogues, e.g. Nei's GST and Weir & Cockerham's θ), and the other is an absolute measure (Nei's genetic identity (I) and genetic distance (D).
BI3010H08 BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow Measures of genetic differentiation: Relative measurel: FST (S.Wright), GST (M. Nei) Absolut measurel: Genetic Identity (I) og Genetic distance (D) (M. Nei). Wright's FST = 1 - (HS/HT), where HS is the mean observed heterozygosity over all subpopulations, HT is the expected heterozygosity i the total materials (based on the allele frequencies in the Total. From the tabulated data, both the subpopulation have an observed (and here also expected) heterozysity of 0.48. The calcuated mean value HS is thus 0.48 as well. The expected (HW) heterozygosity in the Total according to its allele frequencies is: (2*0.5*0.5)=0.5, dvs HT=0.5. Therefore, FST = 1 - (0.48/0.50) = 0.04 in this example. Nei's GST is an expansion of FST to cover multiple alleles and loci. This assumes HW distributions in all subpopulations. Mathematically, Wrigth's and Nei's measures are not principally different. GST can be viewed as a weighted mean of FST over loci.
BI3010H08 BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow An absolute measure of genetic differentiation: Nei's I (Genetic Identity) I = (xiyi) / [ ( (xi2)( (yi2)], where xi, yi is the frequency of the i-th allele in subpopulations X and Y, respecitvely I = (0.6*0.4 + 0.4*0.6) / [ ((0.36 + 0.16)(0.16 + 0.36))] I = 0.48 / 0.52 = 0.9231 Nei's D (Genetic Distance) D = - ln(I) = - ln(0.9231) = 0.08 in this example. This D-values is an absolute measure of differentiation, and stands for an estimate of the mean number og amino acid substitutions ("opposite fixations") per locus. Usually, D-values are calculated as mean values over loci, preferably > 10, and monomorphic as well as polymorphic loci because D shall principally represent a random sample from the genome.
BI3010H08 BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow In a typical study of genetic differentiation between populations within a species, a number of samples are taken from various locations in the species' range. The locations may be picked randomly or according to some underlying hypothesis/theory about structure. At any rate, the genotypic composition and the allele frequencies in each sample are obtained by the pertinent laboratory techniques (usually electroporesis in some form is involved). The next step is usually to calculate some measure of genetic similarity or distance between each sample and all other samples, and to arrange the obtained values in a matrix. The matrix is usually presented graphically after performing: 1. Cluster analyses and 2. Dendrogram construction. In the example on the next pages, a matrix of D-values (Nei 1972) between three populations is subject to cluster analysis and UPGMA* dendrogram construction. A simple case concerning three loci with two alleles (S and F) at each locus is considered. -------------- * UPGMA refers to "Unweighted Paired-Group Method of aritmetic Average"
BI3010H08 BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow Population 1 Population 2 Population 3
BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow Genetic distance (D): The following procedure was originally described by Nei (1972). Nei later published an "unbiased" version of D, which involves a slightly more complex procedure. For the sake of simplicity we shall here use the first one. Formler: I = xiyi / SQR[ (xi2)(yi2)], and D = - ln(I) Calculated values of I and D from observed allele frequencies: Population 1 versus population 2: HbI*:I = (0.5x0.9)+(0.5x0.1) / SQR [(0.25+0.25)x(0.81+0.01)] = 0.781 LDH-3*: I = (0.1x0.5)+(0.9x0.5) / SQR [(0.01+0.81)x(0.25+0.25) = 0.781 IDHP-1*: I = (0.6x0.5)+(0.4x0.5) / SQR [(0.36+0.16)x(0.25+0.25)] = 0.981 -------------------------------------------------------------------------------------------------------------- Mean I = (0.781+0.781+0.981 / 3 = 0.848 Mean D = - ln(0.848) = 0.165 ================================================================
BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow Population 1 versus population 3: HbI*: I = (0.5x0.8)+(0.5x0.2) / SQR [(0.25+0.25)x(0.64+0.04)] = 0.857 LDH-3*: I = (0.1x0.6)+(0.9x0.4) / SQR [(0.01+0.81)x(0.36+0.16)] = 0.643 IDHP-1*: I = (0.6x0.7)+(0.4x0.3) / SQR [(0.36+0.16)x(0.49+0.09)] = 0.983 -------------------------------------------------------------------------------------------------------- Mean I = (0.857+0.643+.983 / 3 = 0.827 Mean D= - ln(0.827) = 0.190 ===================================================================== Population 2 versus population 3: HbI*: I = (0.9x0.8)+(0.1x0.2) / SQR [(0.81+0.01)x(0.64+0.04)] = 0.991 LDH-3*: I = (0.5x0.6)+(0.5x0.4) / SQR [(0.25+0.25)x(0.36+0.16)] = 0.981 IDHP-1*: I = (0.5x0.7)+(0.5x0.3) / SQR [(0.25+0.25)x(0.49+0.09)] = 0.929 --------------------------------------------------------------------------------------------------------- Mean I = (0.991+0.981+0.929 / 3 = 0.967 Mean D= - ln(0.967) = 0.034 ============================================================
BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow CLUSTER ANALYSIS AND DENDROGRAM CONSTRUCTION Matrix 1: Calculated values for I og D (I-values above diagonal and D-values below). Den lowest distance in Matrix 1 is that between population 2 and 3. These are therefore joined together into one OTU (Operational Taxonomic Unit) and are connected by a line at the lowest level in the dendrogram. The matrix is then recalculated, regarding pop.2 and pop3. as one unit, which genetic distance to pop.1 is calculated as the aritmetic mean of pop.1's original distance to each of the two others. In this example this distance is: (0.165 + 0.190) / 2 = 0.178. After this procedure, Matrix 2 looks like on next page.
BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow Matrix 2: D-values below diagonal. This procedure; to join the OTUs showing the lowest D-value in each cyclus and then recalculate the matrix, continues until all OTUs are joined by the bifurcations. In this example there will be two "nodes" (Pop1.1 an the combined Pop.2 and Pop. 3) in a dendrogram based on Matrix 1 and Matrix 2. See the dendrogram on the next page.
BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow 3 3 0 2 2 2 2 "P1" .5 .5 .1 .9 .6 .4 "P2" .9 .1 .5 .5 .5 .5 "P3" .8 .2 .6 .4 .7 .3 ASCII (txt) input file for DG25da.exe UPGMA dendrogram of the genetic distance of Nei (1972) calculated in matrix 1 and 2. Screendump from program DG25da.exe (J. Mork).
BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow Empirical values of Nei's D based om studies on a wide range of taxa at different levels of genetic differentiation.
BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow
BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow
BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow
BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow Cod sampling sites (of Mork et al. 1985)
BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow UPGMA dendrogram, and ’isolation by distance’ for cod (Mork et al. 1985)
BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow There are alternative ways of calculating FST (and its analogues) between populations. As showed previously, the Wahlund effect (and thereby FST) of the "total" increases when the sub-populations become increasingly different. It is therefore logical to use the variance in local allele frequencies relative to the heterozygosity in the total population as a proxy for FST. Wright (1951) first defined the F coefficients as correlations between gametes. He also showed that for two alleles, FST can be expressed using the variance in allele frequencies among subpopulations: FST = V(p) / pmean(1-pmean) The variance V(p) is here calculated as for a continuous variable (i.e. using the mean square method) in n sub-populations. The denominator in the formula uses the mean allele frequency in the sub-populations (cf box 9.5). Weir & Cockerham (1984) defined a parameter θ (theta) which is analoguous to FST; and which can be used for loci with many alleles. It uses the ratio of the variance in allele frequencies among sub-populations to the total variance in allele frequencies. Nei (1973) defined a parameter which is a multi-locus, multi-allelic measure for differentiation which needs no assumptions like absence of selection etc. He called it "the coefficient of gene differentiation", GST. It is closely related to FST. (page 325).
BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow Testin the statistical significance of differentiation Tests are in use which indicates whether a calculated FST is significantly different from zero. They are based on some strict assumptions, e.g. equally sized populations. A general test which can be used in all situations and simultaneously has the highest "power" of them all in detecting differentiation, is the RxC contingency table test (procedure showed in slide 5). An MS DOS program for running the test has been uploaded to It's learning (chiRxC.exe). This test can be used for both genotypic and allelic proportions. (NB! Remember that as in all chi-square tests, counts rather than frequencies must be used). Gene flow and FST Obviously, a gene flow between populations will inhibit differentiation due to genetic drift. The equilibrium between these two opposing forces is described by the relation: FST = 1 / (4Nm+1) The product Nm is the absolute number if immigrants into the local population. While this may seem non-intuitive at first glance, it reflects the fact that the gene flow must balance a larger genetic drift in small populations. If this relation is solved with respect to Nm (formula 9.17), one gets an estimate of the exchange of genetically effective individuals per generation which takes place in the equilibrium situation (which again will be reached after 4N generations). See discussion of this on pages 345 and 364, which warns that this relies on a set of strict assumptions: 1. Pure "Island model" 2. Constant population size 3. N = Ne 4. No mutation 5. Gene flow is random between populations 6. Gene flow is random with respect to genotype 7. No selection at the loci used 8. All populations are in equilibrium with respect to mutation, gene flow, and genetic drift
BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow Assignment tests In principle one can "track" an individual back to its mother population by means of its multilocus genotypic combination. However, this is only possible if one knows all the possible mother populations and their multilocus genotype frequencies. In practice, such knowledge is rarely at hand when studying natural populations.
BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow Summing up chapter 9 1. Population structuring takes place when a large population is divided into several smaller ones, in which the individuals are more disposed to mate with each other than with individuals from another popolation. 2. In a physical mixture of individuals from several populations with different allele- and genotype frequencies, there will be a "Wahlund effect"; a nominal deficiency of heterozygotes compared to HW-ecpected values. 3. The degree of differentiation between subpopulations can be quantified with FST, which is based on the Wahlund-effect. 4. Several analogues ti FST exist. is based on allele frequency variances, GST on "gene diversities". 5. Gene flow is the immigration of individuals or gametes from another population. Gene flow inhibits or prevents genetic differentiation between populations. 6. Many models exist for population structure and gene flow. The "Island model" if often used. 7. All models of gene flow (excluding othe evolutionary forces) converge towards the same, namely that subpopulations will be more similar over time, and allele frequencies will converge towards the average allele frequency for the populations involved.For gene flow from only one donor to one recipient, the recipient will turn to be identical to the donor over a number of generations which depends on immigrant proportion. 8. Gene flow counteracts the effects from the other evolutionary forces (mutations, genetic drift, selection). 9. Generally, very few immigrant individuals per generation are needed to inhibit any substantial genetic differentiation between populations. 10. Differentiation between populations can result from many other factors than restrictions in gene flow; e.g. different selection coefficients, fragmentation, expansion. 11. FST is a good measure of population subdivision, but using it for estimating absolute number of migrants must be done with great caution.
BI3010H08 Population genetics Halliburton chapter 9 Population subdivision and gene flow