Reversible Process
Grains of sand. In limit of infinitesmal changes, system moves through equilibrium states during transition-->reversible process. Reversible Process. Remove one grain of sand at a time. Allow system to equilibrate. Pressure decreases by very small amount.


Reversible Process
E N D
Presentation Transcript
Grains of sand In limit of infinitesmal changes, system moves through equilibrium states during transition-->reversible process Reversible Process Remove one grain of sand at a time. Allow system to equilibrate Pressure decreases by very small amount Volume increases by very small amount. A reversible process is one where every step of the path is at equilibrium with its surroundings
Examples of Reversible and Irreversible • Jumping into a pool? (from the perspective of the water) • Getting into a pool slowly? • Free expansion of a gas • Isothermal expansion (piston must move very slowly for bath) • Moving a glass slowly
Isothermal Reversible Process: T constant: Ideal gas For an ideal gas U= q + w =0 -w = q
Isothermal Reversible Process: T constant: Ideal gas For an ideal gas E=q+w=0 w=-q In rev process Pext=Pgas=P=nRT/V w=-P1V -P2V- …
Free Expansion the same final state the same initial state Internal energy of an ideal gas Partition is broken and gas is Allowed to expand in an adiabatic (no heat transfer container) Controlled expansion (Isothermal Reversible) Work done No work done Different paths Heat exchanged No heat exchanged Is DU is the same for both processes?
Different paths in the Expansion of a gas Consider the expansion of an ideal gas with various coupling between it (the system) and the surroundings • Free expansion (No external Pressure) • Isothermal (Constant Temperature) • Adiabatic (No heat transfer) • Isobaric (Constant pressure) • Isochoric (No work) Thermal reservoir
Change in State at Constant Volume 1) 2) Substitute in 1) into 2) What is this at constant volume? How do we measure heat flow? (calorimeter) What is for an Ideal Gas?
Free expansion Joule originally did a free expansion experiment and found that virtually no heat was transferred between the system and its surroundings. • U = KE + PE • U = q + w • w= pextdV , pext=0, w=0 • No temperature change in surroundings so q=0 • Therefore DU =0, the internal energy is independent of pressure and volume and is only dependent on Temperature This is a calorimeter
Joule originally did a free expansion experiment and concluded that virtually no heat was transferred. However, for non-ideal gas some temperature change occur. The internal energy U is the sum of the kinetic and potential energies for all the particles that make up the system. Non-ideal gas have attractive intermolecular forces. So if the internal energy is constant, the kinetic energies generally decrease (think of the vanderwaals potential). Therefore as the temperature is directly related to molecular kinetic energy, for a non-ideal dilute gas a free expansion results in a drop in temperature. U(r) Internal energy of Real Gas r van der Waals interaction What about for a dense gas?
Joule did his experiments with relatively dilute gases so that their behavior was similar to an ideal gas. How would you expect a denser gas to behave? When thinking about this, think of the Vanderwaals attractions between the particles. Would they become more or less favorable? If the expansion was done in an adiabatic container what would happen? Would the final temperature be higher, lower or remain the same? If the Temperature was controlled with a bath would heat be transferred into or out of the system?
Free Expansion the same final state the same initial state Internal energy of an ideal gas Partition is broken and gas is Allowed to expand in an adiabatic (no heat transfer container) Controlled expansion (Isothermal Reversible) Work done No work done Different paths Heat exchanged No heat exchanged Is DU is the same for both processes?
Problem Imagine that an ideal monatomic gas is taken from its initial state A to state B by an isothermal process, from B to C by an isobaric process, and from C back to its initial state A by an isochoric process. Fill in the signs of Q, W, and ¢U for each step. P, A 2 0 T=const B 1 C 1 2 V
Problem Imagine that an ideal monatomic gas is taken from its initial state A to state B by an isothermal process, from B to C by an isobaric process, and from C back to its initial state A by an isochoric process. Fill in the signs of Q, W, and ¢U for each step. P A 2 + -- 0 T=const -- + -- B 1 C + 0 + 1 2 V Is this an engine?
heat QI heat QII C) QI > QII A) QI < QII B) QI = QII Two other Systems • Consider the two systems shown to the right. In Case I, the gas is heated at constant volume; in Case II, the gas is heated at constant pressure. Compare QI , the amount of heat needed to raise the temperature 1K (= 1ºC) insystem I to QII, the amount of heat needed to raise the temperature 1ºC insystem II. TexPoint fonts used in EMF. Read the TexPoint manual before you delete this box.: AAAA
heat QI heat QII C) QI > QII A) QI < QII B) QI = QII U = Q - Wby ACT 3 Solution • Consider the two systems shown to the right. In Case I, the gas is heated at constant volume; in Case II, the gas is heated atconstant pressure. Compare QI , the amount of heat needed to raise the temperature 1ºC insystem I to QII, the amount of heat needed to raise the temperature 1ºC insystem II. • Apply the First Law of Thermodynamics to these situations! • In Case I, NO WORK IS DONE, since the volume does not change. • In Case II, POSITIVE WORK IS DONE by the system, since the volume is expanding, therefore Wby > 0 • In each case, temperature rise is the same, and so is DU. Therefore, more heat is required in Case II!
Change in State at Constant Volume 1) 2) Substitute in 1) into 2) What is this at constant volume? What do each of these terms mean? How do we measure heat flow? (calorimeter) What is for an Ideal Gas? For a Lennard Jones Gas?
Heat Capacity at Constant Volume The heat capacity at constant volume CV is defined as: It takes more energy to increase the temperature of a material that has a higher heat capacity. Heat and Heat Capacity are both extensive properties. That is, they depend on the amount of material present. We therefore define Molar Heat Capacity = heat capacity per mole; Cm = C/N Specific Heat = heat capacity per gram; c = C/mass. For an ideal gas,
The internal energy for an ideal gas is just dependent on the Temperature so both states at T2 have the same Energy At constant-volume (no work is done) At constant-pressure (work is also done) Heat capacity of an ideal gas at constant Pressure
The Enthalpy Nice to have a quantity for Isobaric processes (P = const): H U + PV - the enthalpy This is a Legendre Transform of the Energy At constant Pressure the change in enthalpy is equal to the heat transferred during the process:
The Enthalpy Nice to have a quantity for Isobaric processes (P = const): H U + PV - the enthalpy The enthalpy is a state function, because U, P, and V are state functions. In isobaric processes, the energy received by a system by heating equals to the change in enthalpy. in both cases, Q does not depend on the path from 1 to 2. isochoric: q = U isobaric: q = H Consequence: the energy released (absorbed) in chemical reactions at constant volume (pressure) depends only on the initial and final states of a system. The enthalpy of an ideal gas: (depends on T only)
CV and CP the heat capacity at constant volume V = const P = const the heat capacity at constant pressure To find CP and CV, we need f (P,V,T) = 0 and U = U (V,T)
CV and CP the heat capacity at constant volume V = const P = const the heat capacity at constant pressure To find CP and CV, we need f (P,V,T) = 0 and U = U (V,T)
= U 3 Nk T Internal Energy of a Classical ideal gas • “Classical” means Equipartition Principle applies: each molecule has average energy ½ kT per quadratic modein thermal equilibrium. At room temperature, for most gases: • monatomic gas (He, Ne, Ar, …) • 3 translational modes (x, y, z) U= 3/2 NkT diatomic molecules (N2, O2, CO, …) 3 translational modes (x, y, z) + 2 rotational modes (wx, wy) U= 5/2 NkT • non-linear molecules (H2O, NH3, …) • 3 translational modes (x, y, z) • + 3 rotational modes (wx, wy, wz) • For an ideal gas, the internal energy only depends on the Temperature: U=® NkT = ® nRT • ( f depends on the type of molecule)
CV and CPforan Ideal Gas For an ideal gas CV = dU/dT CVof one mole of H2 7/2NkB Vibration 5/2NkB Rotation 3/2NkB Translation # of moles 100 1000 10 T, K 0 ( for one mole ) For one mole of a monatomic ideal gas:
E depends only on T Constant Volume Heat Capacity for monoatomic ideal gas
E depends only on T Constant Volume Heat Capacity for Diatomic Ideal Gas
Quasistatic Processes in an Ideal Gas • isochoric ( V = const) P 2 PV= NkBT2 1 PV= NkBT1 V1,2 V • isobaric ( P = const ) P 2 1 PV= NkBT2 PV= NkBT1 V1 V2 V
Grains of sand In limit of infinitesmal changes, system moves through equilibrium states during transition-->reversible process Reversible Process Remove one grain of sand at a time. Allow system to equilibrate Pressure decreases by very small amount Volume increases by very small amount. A reversible process is one where every step of the path is at equilibrium with its surroundings
Thermodynamic processes of an ideal gas(DU = q - wby ) 2 p Q Q 1 Temperature changes FLT: V p • Isobaric (constantpressure) 1 2 p FLT: Temperature and volume change V • Isochoric (constant volume)
1 2 p Q Thermal Reservoir T FLT: V Volume and pressure change • Adiabatic (isolated - no heat input) 1 2 p FLT: Volume, pressure and temperature change V ( FLT: DU = q + w ) • Isothermal (constant temperature)
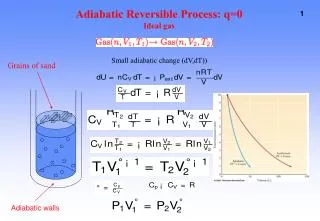
Details of an Adiabatic process (Q = 0) U, W for an ideal gas: Know this derivation. Integrate: TaV = constant Using pV = NkT, we can also write this in the form: where = (+1)/ pVg = constant
isotherm : pV = constant • In an adiabatic process, p, V, and T all change monatomic gas g =(5/2)/(3/2) = 5/3 = 1.67 diatomic gas g =(7/2)/(5/2) = 7/5 = 1.4 • Adiabats (adiabatic p-V curves) are steeper than isotherms • Adiabatic and isothermal processes are reversible -- no heat flow between objects at different temperatures. The p-V curve for an Adiabatic process adiabat : pVg = constant = (+1)/ p V
Reversible Processes A reversible process is at equilibrium (mechanical and thermal) with its surroundings at all times. The path is always on the surface described by the equation of state.
Examples of Reversible and Irreversible • Jumping into a pool? (from the perspective of the water) • Getting into a pool slowly? • Free expansion of a gas • Isothermal expansion (piston must move very slowly for bath) • Moving a glass slowly
Isothermal Reversible Process: T constant: Ideal gas For an ideal gas \Delta U= q + w =0 -w = q
Isothermal Reversible Process: T constant: Ideal gas For an ideal gas E=q+w=0 w=-q In rev process Pext=Pgas=P=nRT/V w=-P1V -P2V- …
Isothermal Process in an Ideal Gas P • isothermal ( T = const ) : PV= NkBT W V2 V1 V Wi-f > 0 if Vi>Vf (compression) Wi-f < 0 if Vi<Vf (expansion)
Adiabatic Process in an Ideal Gas • adiabatic(thermallyisolated system) The amount of work needed to change the state of a thermally isolated system depends only on the initial and final states and not on the intermediate states. P 2 to calculate W1-2 , we need to know P(V,T) for an adiabatic process PV= NkBT2 1 PV= NkBT1 V2 V1 V ( f – the # of “unfrozen” degrees of freedom )
Adiabatic Process in an Ideal Gas (cont.) P 2 An adiabata is “steeper” than an isotherma: in an adiabatic process, the work flowing out of the gas comes at the expense of its thermal energy its temperature will decrease. PV= NkBT2 1 PV= NkBT1 V2 V1 V 1+2/31.67 (monatomic), 1+2/5 =1.4 (diatomic), 1+2/6 1.33 (polyatomic) (again, neglecting the vibrational degrees of freedom)
Problem Imagine that we rapidly compress a sample of air whose initial pressure is 105 Pa and temperature is 220C (= 295 K) to a volume that is a quarter of its original volume (e.g., pumping bike’s tire). What is its final temperature? Rapid compression – approx. adiabatic, no time for the energy exchange with the environment due to thermal conductivity For adiabatic processes: also - poor approx. for a bike pump, works better for diesel engines
Isothermal Reversible Change • In an isothermal reversible expansion, temperature does not change • T is not a function of V and can be removed from the integral • For ideal gas, w = -nRT ln (Vf/Vi) • If final volume > initial (expansion), w < 0 • System has done work on surroundings internal energy(U) decreased • If final volume < initial (compression), w > 0 • System has work done on it, U increased • Note that as T increases |w| increases
Reversible vs. Irreversible Change • Irreversible expansion: w= = -pexDV • Reversible (isothermal) expansion: w=-nRTln{Vf/Vi} • Which is greater? • Consider indicator diagram • Work(rev.) = Area under curve • Work (irrev.) = Area under rectangle • Work (rev.) > W (irrev.) • Reversible work is the maximum which can be done • True of PV work • True of all work
Heat Changes - Calorimetry • Recall: dU = dq + dw • If expansion work is dwexp and dwother is other work done (electrical, magnetic, etc.), then dw = dwexp + dwother • dU = dq + dwexp + dwother • If V is constant, dwexp is zero since no PV work can be done • Assume dwother is also zero, then dU = dq = qv • qv is the change in q at constant volume • Thus, measuring the heat change in a system at constant volume gives a measure of the change in internal energy • This process of measuring heat change is known as calorimetry • One constant volume container for measuring heat change is a bomb calorimeter • Experiment 1 in lab • Typically, substance is burned in calorimeter and temperature rise is measured (dV is constant, but P does change in bomb) • In a bomb calorimeter, DT a qv • This proportionality is quantified by calibration, typically by combusting a known substance
Heat Changes - Heat Capacity • If V constant, U increases as temperature increases • The rate of change of U at any temperature, (dU/dT)V is called the heat capacity, CV • CV(A) is not equal to Cv(B) generally smaller if TA < TB • Note volume is const. If that changes CV(T may change) • CV is an extensive property (2x the amt gives twice the heat capacity) • Molar heat capacity, CVm, is the • Rate of change of CV with T typically small around room temperature, you can assume it’s constant • This means that dU = qV= CV dT ≈ CVDT • You can estimate CV by determining the amount of heat supplied to a sample • Because qV ≈ CVDT, for a given amount of heat the larger CV, the smaller DT • At a phase transition (boiling point) DT = 0 so CV = ∞
Grains of sand In limit of infinitesmal changes, system moves through equilibrium states during transition-->reversible process Reversible Process Remove one grain of sand at a time. Allow system to equilibrate Pressure decreases by very small amount Volume increases by very small amount. A reversible process is one where every step of the path is at equilibrium with its surroundings
Examples of Reversible and Irreversible • Jumping into a pool? (from the perspective of the water) • Getting into a pool slowly? • Free expansion of a gas • Isothermal expansion (piston must move very slowly for bath) • Moving a glass slowly
Isothermal Reversible Process: T constant: Ideal gas For an ideal gas \Delta U= q + w =0 -w = q
Isothermal Reversible Process: T constant: Ideal gas For an ideal gas E=q+w=0 w=-q In rev process Pext=Pgas=P=nRT/V w=-P1V -P2V- …
¢ U depends only on ¢T Constant Volume Heat Capacity for monoatomic ideal gas
E depends only on T Constant Volume Heat Capacity for Diatomic Ideal Gas