Pharmaceutical Issues
Pharmaceutical Issues. Shanker Gupta, Ph.D. Pharmaceutical Resources Branch (PRB). Requirements. Chemical Substance to Human Drug Standardize Processes to Establish Identity Quality Purity Strength. Operational Areas. Bulk Drug Synthesis Analytical Development & Validation


Pharmaceutical Issues
E N D
Presentation Transcript
Pharmaceutical Issues Shanker Gupta, Ph.D. Pharmaceutical Resources Branch (PRB)
Requirements Chemical Substance to Human Drug • Standardize Processes to Establish • Identity • Quality • Purity • Strength
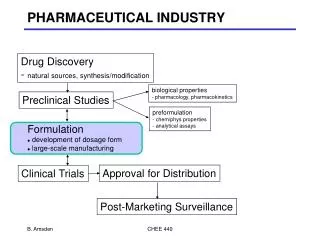
Operational Areas • Bulk Drug Synthesis • Analytical Development & Validation • Formulation Research & Development • Clinical Dosage Form Manufacture • Shelf Life Monitoring
Research Contracts • Solicitation - RFP Process • Select Organizations with Expertise • Selected by Open Competition • Contracts Awarded for 5-year Periods
PRB Staff • Rao Vishnuvajjala, Ph.D., Chief • Shanker Gupta, Ph.D. • Sung Kim, Ph.D. • Paul Liu, Ph.D. • Ken Snader, Ph.D.
Bulk Drug Synthesis • Develop Synthetic Route • Purification Process • Scale Up of Reactions • Improve Purification Process & Optimize Yield • Develop GMP Procedures • Prepare Large Batches & Provide Documentation • Prepare Reference Standard
Synthetic Projects SarCNU DB-67 RH-1 CDDO
Synthetic Projects PT-523 5-Azacytidine BPU 17-AAG
Analytical Chemistry (general) • Identity of Molecular Structure • IR, NMR, MS, EA, etc… • Develop Purity Tests • HPLC, GC, TLC, etc… • Test for Other Impurities • Moisture (K-F), Residual Solvents (GC) • ROI, Heavy Metals
Analytical Chemistry (batches) • Confirm Identity • Determine Purity & Potency (assay) • Levels of Other Impurities • Methods Validation • Establish Release Specifications • Stability of Bulk Drug Substance
Typical Specifications • Bulk Drug Substance • Purity: > 98% • Impurities: NMT 2% total; 0.5% single • Residual Solvents: NMT 0.1 to 0.2% • Residue on Ignition: NMT 0.1%
Formulation Research (Goals) Provide Solution of Active Drug for IV Use. Solution components should be: • Compatible with Blood • Non-Toxic to Blood Components and Host • Should be Free of Particulates • Isotonic • Suitable for Sterilization • Approved, USP/NF (when possible) • Proven Safety Record
Solubility Options • Salt Formation • Co-Solvents: DMSO, EtOH, PG, PEG,etc. • Surfactants: Cremophor EL, Tweens, etc. • Emulsions: O/W, EPL, Pluronics, etc.. • Complexes: HPCD, SBE-4, Counter Ionic. • Pro-Drugs • Liposome
Stability Options • Hydrolysis Lyophilize • Oxidation Anti-Oxidants • Photochemical Amber Vials, Boxes • Thermal Storage Temp.
Formulation Research • Defines • Components & Composition • Type of Dosage Form • Container/Closure System • Storage Conditions • Reconstitution Vehicles (Lyo products) • Useable Periods • Stability and Compatibility with Devices
Liposomal Formulation of DB-67 • A liposomal formulation was proposed containing DMPC. DMPG and the drug in a molar ratio of 1:30 (drug:lipid). • The formulation procedure used organic solvents that had to be evaporated with the film suspended in phosphate buffer containing sucrose. • The resulting suspension was fairly coarse with relatively large particle size. The particle size was reduced by extruding the suspension through a polycarbonate membrane.
Scale Up Considerations • It was not possible to use certain organic solvents due to safety considerations. • The drug is fairly lipophilic and was lost in the polycarbonate membrane owing to adsorption. • It is not possible to use polycarbonate membranes readily in a large scale process. • The suspension at the end of extrusion was not reproducibly reduced in particle size such that it can be sterilized by membrane filtration. • It is obvious that this process cannot be validated for GMP production of a human phase I clinical batch without modifications.
Refined Process • The procedure was modified by eliminating the extrusion step in favor of microfluidization. This procedure reduces particle size by applying high energy shear forces to the suspension. • This also resulted in avoiding drug losses due to adsorption to the polycarbonate membrane. • The particle size at the end of the homogenization step was reproducibly less than 0.22 microns such that the product can be sterilized by membrane filtration.
Refined Process for Clinical Batch • At the end of sterilization, the drug suspension was filled in vials and lyophilized. • A conservative lyophilization cycle was used. The suspension was frozen, and lyophilization in a multi-step drying process such that the resulting film (next slide) could be easily rehydrated using 2 mL of WFI yielding 1 mg/mL of drug suspension suitable for injection.
Drug Product Manufacturing • Manufacture of Pilot Batches • Accelerated Stability Testing • Raw Material Qualifications • Bio-Load Determination • Microbial Validations • Preparation of PIP • Preparation of Master Batch Record • Design and Printing of Labels
Drug Product Manufacturing • Follow GMP and QC Procedures • Follow all SOPs • Prepare Drug Product, Sterilize, and Lyophilize as Applicable • Quality Control Testing, Including Chemical & Microbial Testing • Establish Product Release Specifications • QC Release, Labeling, and Packaging
Drug Product Manufacturing • QC/QA Review of Procedures and Batch Record • Ship Batches to NCI Repository • Provide Documentation for IND • Special Studies: • Solution Stability and Compatibility with Infusion Solutions and Devices (e.g., pumps, bags, iv sets, etc.)
Shelf Life Monitoring • Develop Storage & Stability Testing Protocols • Monitor Stability on Established Time Schedule • Promptly Recall Batches that Fall Outside Established Specifications • Each Clinical Lot is Monitored for Stability
Our next speaker is: Dr. Joseph Covey Toxicology & Pharmacology Branch Developmental Therapeutics Program