What are mitochondria?
170 likes | 388 Views
Describe the mitochondrial genome and its major features and review techniques employed in the genetics laboratory for the analysis of mitochondrial disease. What are mitochondria?. Semiautonomous cellular organelles numbers range from 2 to 100’s depending cell type

What are mitochondria?
E N D
Presentation Transcript
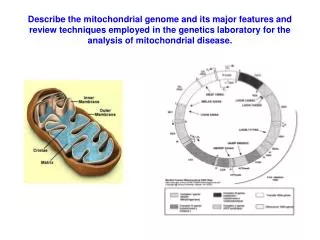
Describe the mitochondrial genome and its major features and review techniques employed in the genetics laboratory for the analysis of mitochondrial disease.
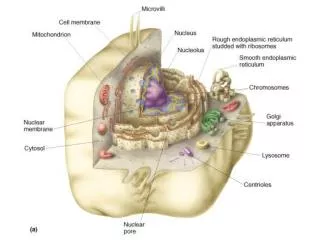
What are mitochondria? • Semiautonomous cellular organelles • numbers range from 2 to 100’s depending cell type • Thought to possibly have started life as bacteria that invaded a nucleated cell • Power-houses of aerobic eukaryotic cells • They contain the electron transport chain which transfers electrons to oxygen by means of a process called oxidativephosphorylation. • releases energy for the production of ATP by forming a pH and electrical gradient across the inner mitochondrial membrane
Mitochondrial genome, Major features 1 • Mitochondrial genome is 16,560bp • 44% GC • Double stranded circular • Two strands very different in sequence • Heavy strand = Rich in guanines • Light strand rich in cytosines • Compact, encodes only 37 genes • 2 = Ribosomal RNAs (Heavy strand) • 22 = tRNAs (14 Heavy, 8 Light) • 13 = polypeptides, all components of the respiratory chain/oxidative phosphorylation system. (12 Heavy, 1 Light) • Maternal inheritance • Oocyte mtDNA from mother to offspring • No paternal contribution (generally although there are some suggestions that this may occur very very rarely)
Transcription and replication • Transcription of H and L strands is from promoters in the triple stranded, non-coding D-loop region of the genome • Transcription from Heavy strand is CLOCKWISE • Transcription from Light strand is ANTICLOCKWISE • No introns, genes separated by 1 or 2 non-coding bases • Multigenic transcripts generated then cleaved • H-strand origin of replication is in D-loop • Replication of H-strand involves using the L-strand as a template and displacing the old H strand. • L-strand replication starts after 2/3 of daughter H-strand has been replicated • L-strand replication proceeds in the opposite direction using the H strand as a template
Mitochondrial genetic code • Mitochondrial genome encodes all rRNA and tRNA molecules it needs for protein synthesis • Need nuclear encoded genes to provide all other components • Mitochondrial genetic code has drifted from the universal code due to small functional load • 60 sense codons, not 61 • 4 stop codons for instead of three • Two Trp codons instead of one • Four Arg codons instead of six • Two Met codons instead of one • Two Ile instead of three • Genetic wobble hypothesis means 22 tRNA species can code for 60 codons
Mitochondrial disease • Definition: Heterogeneous group of disorders that arise as a result of dysfunction of the mitochondrial respiratory chain • Mitochondrial disease can be caused by: • Deletions / duplications in the mitochondrial genome • Kearns Sayre syndrome • CPEO (Chronic progressive external opthalmoplegia) • Pearsons syndrome • Point mutations in the mitochondrial genome • MELAS • MERF • Missense mutations in nuclear DNA altering the function of proteins in the respiratory chain. • Leigh syndrome • Encephalopathy
Major features 2 • Mitochondrial genome is polyploid • Each cell has 100’s of mitochondria with several copies of mtDNA • In normal conditions individuals have one species of mtDNA • HOMOPLASMY • Diseased individuals can have a mixture of mutant and wild type mtDNA • HETERPLASMY • Varied clinical phenotype with individuals within the same family probably due to heteroplasmy • There is a critical threshold of mutant mtDNA before disease is seen • A heteroplasmic female may transmit variable amount of mutant mtDNA to offspring due ‘bottle neck’ hypothesis
Mutability • Mitochondrial genome has a high mutation rate (approximately 10 times higher than nuclear DNA) • They do not have an efficient repair system • They lack protective HISTONES • The genome is physically associated with the inner mitochondrial membrane, so is in close proximity to highly mutagenic oxygen radicals. • No introns so any mutation is likely to affect the codon region • Mutations can be sporadic or maternally inherited • Mutations can be detected in the mitochondrial genome to confirm diagnosis of mitochondrial disease • Sample type important (muscle biopsy rather than blood is preferred for many mutations) • Rearrangements not readily detectable in blood of adults • MELAS mutation 3243A>G may be missed in blood of older patients
Testing for point mutationsin mtDNA • Targeted mutation analysis or genome scanning • e.g. RFLP, SSCP, DGGE, sequencing, WAVE-dHLCP (Transgenomic kit) • Problems encountered can include: • Reporting levels of heteroplasmy • Low, medium, high? • ? Use quantitative methods such as real time PCR, pyrosequencing or known controls with restriction digests • Detecting low level heteroplasmic variants • Sequencing will not detect mutations at heteroplasmic levels of less than 30% • Transgenomic kit reports detection down to 0.5%!! • Variable phenotype with the same mutations causing different diseases. E.g. A3242G can cause MELAS, Cardiomyopathy and Diabetes and Deafness • Whole genome screen • Target tests according clinical information provided
Testing for large scale rearrangements in mtDNA 1 • Techniques: • Long range PCR • Provides rapid reliable exclusion of rearrangements • Artefacts eliminated by DNA dilution • Rearrangements confirmed by second technique (Southern blotting) • Can detect levels of rearrangements that are not clinically significant • Southern blotting • Digestion of DNA with SnaB1 to detect duplications • Digestion of DNA with PvuII or BamH1 to detect deletions • Can also be used to detect depletion disorders • Time consuming
Real time PCR Specialised equipment MLPA? MRC Holland kit exists, however, No control probes Changes of less than 50% decrease or increase in probe signal expected Deletions / duplications probably only detected when more that one probe is located within the area Testing for large scale rearrangements in mtDNA 2 Testing for point mutations in the nuclear genome • Targeted mutation analysis or genome scanning • SURF1, COX10, SCO2, COX15, POLG , ANT1, Twinkle etc
Risks to offspring • mtDNA deletions generally occur de novo, recurrence risk is low (1/24) • mtDNA point mutations and duplications may be transmitted through the maternal line • All offspring of females with a mtDNA mutation are at risk ofinheriting the mutation (variable amounts of mutant mtDNA) • Offspring of males with mutant mtDNA are not at risk • Offspring of patients with nuclear gene mutations are either: • Heterozygotes (autosomal recessive disease) • At 50% risk of inheriting the mutation (autosomal dominant)
Prenatal diagnosis 1 • mtDNA mutations difficult because of mtDNA heteroplasmy • % mtDNA in CVS may not reflect level in other fetal tissue • Mt DNA levels may change during development and life • Therefore for most mtDNA mutations prenatal diagnosis is not recommended • Mutations T8993G/C that cause NARP and MILS does not appear to change significantly over time • Successful prenatal diagnosis has been carried for these two mutations • Relationship between mutant load in mother and disease in offspring for MERF 8344 A>G • Mutant levels below 40% in mother rarely give rise to severe disease in offspring • CVS offered to mothers with mutant load of more than 40%
Prenatal diagnosis 2 • Prenatal diagnosis of MELAS (3243A>G) is not indicated • Severity is not clearly related to mutant load • Prenatal diagnosis recommended in patients with Kearns Sayre syndrome where rearrangement is detectable in blood at >5% • Prenatal diagnosis not usually recommended in other deletion / syndromes • Prenatal diagnosis of mutations in nuclear genes causing mitochondrial disease is possible providing the disease causing mutation has previously been identified