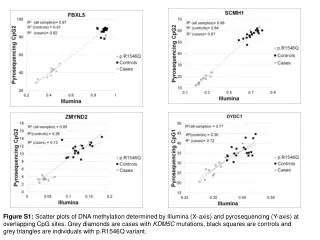
Download

1 / 23
260 likes | 585 Views
How can aberrant DNA methylation give rise to disease. 09/11/2009 Helen Lord. What is methylation. Cytosine methylation is the most common DNA modification in vertebrates Only ~3% of cytosines in human DNA are methylated Most of this occurs at CpG dinucleotides

E N D
How can aberrant DNA methylation give rise to disease 09/11/2009 Helen Lord
What is methylation Cytosine methylation is the most common DNA modification in vertebrates Only ~3% of cytosines in human DNA are methylated Most of this occurs at CpG dinucleotides Addition of a methyl group to the C5 position of this dinucleotide creating 5-methylcytosine Catalysed by: De novo Methyltransferases – expressed mainly in early embryo development and these set up the pattern of methylation Maintenance Methyltransferases
Process of methylation Methylation by DNA methyltransferases at CpG islands – uses S-adenosylmethionine (SAM) as the methyl donor (results in S-adenosylhomocytosine) DNA demethylation relaxes chromatin structure allowing histone acetylation and the binding of transcriptional complexes.
What is the function of DNA methylation • DNA methylation is a mechanism for silencing and transcription. • Regulating expression of endogenous genes and reducing transcriptional noise. • This reduction in unnecessary gene expression may have permitted the increase in gene number and in complexity that characterises vertebrates. • Whilst DNA methylation at CpG islands downstream of promoters does not block controlled transcription through these regions, there is no doubt that methylated promoter regions correlate with transcriptional silencing. • Critical role in cellular processes including embryonic development, transcription, X chromosome inactivation and genomic imprinting.
How does aberrant DNA methylation give rise to disease In normal cells DNA methylation occurs predominantly in repetitive genomic regions Including satellite DNA and parasitic elements (such as long interspersed transposable elements (LINES) and short interspersed transposable elements (SINES)). CpG islands, particularly those associated with promoters are generally unmethylated although an increasing number of exceptions are being identified. Normal methylation helps to maintain the genomic stability and prevent illegitimate recombination. Aberrant hypomethylation causes disease by: Allowing transcription of genes that should be silenced (use of unmasked promoters) Generation of antisense transcripts through using unmasked promoters will therefore lead to silencing of genes that should be active Genomic instability – hypomethylated repetitive regions are more prone to recombination
How does aberrant DNA methylation give rise to disease Aberrant methylation at CpG loci CpG islands around promoters are generally unmethylated Some exceptions – some promoters are methylated in an allele specific manner = DMR’s (differently methylated regions) therefore the dosage of these regions is important for development (Imprinted genes) Aberrant methylation of imprinted genes leads to disease by changing the dose of the gene products -hyper/hypomethylation. Causes of aberrant methylation to imprinted loci are usually due to: imprinting control region mutations (ICR) – PWS/AS Uniparental disomy (UPD) - RSS Deletion/duplication of one allele which alters the dosage of the gene product.
Example 1- Variants in proteins involved in methylation • Rett syndrome • Neurological disorder caused by mutations in the MECP2 (methyl CpG binding protein 2) gene. • MeCP2 protein binds specifically methylated DNA sequences – acts as a transcriptional repressor. • MeCP2 can also bind to RNA in vitro and might influence RNA splicing in vivo. • Highly expressed, but at relatively high levels in the brain. This distribution may explain the phenotype seen (MeCP2 increases in the postnatal period of neuronal maturation). • ICF syndrome • Primarily an immunodeficiency characterised by agammaglobulinaemia with B cells. • Approx half patients = mutations in DNMT3B. Don’t fully affect the gene function but you see regional hypomethylation. • Mice studies showed LOF = deregulation of important developmental regulators critical for development of the immune system, brain and craniofacial development.
Example 2- Imprinting disorders D15S18 D15S817 D15S122 D15S540 15SCA-2 D15S219 D15S511 D15S24 Imprinting centre Telomere Centromere MKRN3 SNURF HERC2 MAGEL2 SNRPN P (OCA2) GABRG3 NDN PAR5 GABRA5 IPN PAR1 GABRB3 UBE3A ATP10C Key Paternally expressed genes Imprinting centre Maternally expressed genes AS-SRO PWS-SRO Other genes located in the PWS/ AS region • Imprinted genes are expressed in a parent of origin manner, therefore show monoallelic expression – other allele is silenced via CpG island methylation in the promoter region. • PWS/AS imprinted region Defects in oppositely imprinted genes on chromosome 15 • PWS – caused by loss of paternal unmethylated allele at the PWS ICR. • Loss of transcription of many genes in the 15q11-q13 region – ZNF127,SNURF/SNRPN and NDN. • AS – Caused by loss of maternally expressed UBE3A gene. • Other examples: Beckwith Wiedemann syndrome (BWS), Russell-Silver syndrome (RSS) and transient neonatal diabetes mellitus (TNDM).
Example 3 - Methylation in the triplet repeat disorders Fragile X syndrome CCG expansion in the 5’UTR of the FMR1 gene seen in patients with fragile X syndrome. In normal and premutation individuals the promoter region of FMR1 is unmethylated (<200 rpts) In individuals with >200 rpts the promoter region becomes methylated (euchromatin > heterochromatin) leading to transcriptional silencing of FMR1. Myotonic dystrophy (DM1) Multisystem disorder caused by expansions of the CTG repeat in the 3’ UTR of the DMPK gene. At the epigenetic level : normal alleles - the repeat is organised as heterochromatin, incl. repressive histone modifications and bidirectional modification (restricted to the repeat sequence by insulator elements either side of the repeat). At the mutant DM1 locus the insulator function is lost therefore spread of heterochromatin in euchromatic domains flanking the repeat.
Example 4 – Methylation in microsatellite repeat disorders FSHD (Facioscapulohumeral muscular dystrophy) AD FSHD is caused by a contraction of the polymorphic microsatellite repeat D4Z4 in the subtelomere of chromosome 4q. This repeat consists of KpnI units of 3.3kb – vary between 11 and 100 repeat elements in a single array. Patients with FSHD almost always have one contracted allele of 1-10 units. Exact mechanism poorly understood, however, DNA methylation studies suggest a prominent epigenetic disease mechanism. D4Z4 repeats are normally highly methylated but in patients with FSHD disease repeats are hypomethylated. Also a small proportion of patients without D4Z4 repeat contraction show the same loss of D4Z4 methylation.
Example 5 – Methylation and cancer First observed in colorectal cancer Genomes of cancer cells are often hypomethylated in the repetitive regions of the genome lead to chromosome instability and inappropriate gene activation. Genes involved in cell cycle regulation and DNA repair often have hypermethylation at their promoters causing the gene to be silenced (often the first hit). E.g. Hypermethylation of the hMLH1 promoter in HNPCC E.g. Mutations in p16 can lead to melanoma, however p16 promoter methylation and gene silencing is seen in several cancers incl. Small cell lung carcinoma, breast cancer, and is thought to be an early event in tumour development.
Example 5 – Methylation and cancer Loss of Imprinting and cancer (LOI) When a silent allele in growth promoting imprinted genes is activated this increases the expression of a gene product therefore leading to a growth advantage. E.g. Wilms tumour - LOI at the IGF2 / H19 locus. When the active allele of a growth inhibitory gene is silenced – may cause deregulation of cell growth E.g. In some Wilms tumour patients - LOI at the P57KIP2 locus NB patients with BWS have an 800 fold increased risk of developing Wilms tumour of the kidney and rhabdomyosarcoma.
Methods for detection – Methylation-sensitive restriction enzyme analysis This is the traditional method used for identifying methylation status Involves a double digest of both a methylation sensitive and a methylation insensitive enzymes Methylation sensitive enzyme cuts in different places depending on the methylation status of each strand Seperation of digested products by electrophoresis Southern blotting is undertaken using a probe specific to the area of interest This can be done either radioactively or by chemiluminescence. Size of methylated and unmethylated digest products differ, therefore methylation status can be identified based on size.
Methods for detection – Methylation-sensitive restriction enzyme analysis • Chemiluminescence blotting: non-radioactive labelled probe • The probe is labelled with digoxigenin (Dig) by either end-labelling, random-primed labelling and PCR incorporation (the latter is best). • Anti-Dig Ab coupled to Alkaline Phosphatase binds to the dig, and this acts on an added Chemiluminescence substrate (CDP-Star) producing a light signal. • Advantageous as no heath risks for user • Higher sensitivity (compared to radioactivity) • Small amount of template required • Faster results compared to isotopic procedures • especially at the detection stage (~20 mins • compared to o/n-weeks) • Stable probe • Flexible – easy to describe bespoke probes • Can use impure templates
Methods for detection – Bisulfite treatment of DNA Treatment of single stranded DNA with sodium bisulfite – this selectively deaminates cytosine residues and converts them to uracils (methylated cytosines are protected). The modified sequence can then be analysed for evidence of sequence variations by using a range of PCR amplification based methods. Converted DNA is amplified using primers that do not differentiate between methylated and unmethylated sequences. Direct sequencing - methylated cytosine residues are retained in the resulting sequence, whereas unmethylated cytosines are converted to uracils and will therefore appear as thymine residues. COBRA (combined bisulfite restriction analysis) DNA is amplified as before, but then subjected to digestion with sequence specific restriction enzymes whose DNA recognition sites recognise CpG residues. MSP (Methylation sensitive PCR) the primers used specifically recognise and amplify either methylated or unmethylated target DNA sequences. The DNA is amplified in parallel with the two primer sets and PCR products can be analysed by gel electrophoresis of fluorescently.
Methods for detection – Bisulfite treatment of DNA Diagram showing three different ways of analysing bisulfite treated DNA as described on the previous slide.
Methods for detection – Bisulfite treatment of DNA - ABI traces I – AS patient – The presence of the paternal (unmethylated) PCR product at 216bp, and the absence of the maternal (methylated) PCR product at 313bp = complete loss of maternal methylation. II – Healthy control The presence of both paternal (unmethylated) and maternal (methylated) PCR products at 216bp and 313 bp = percentage of methylation consistent with a normal profile. III – PWS patient - The absence of the paternal (unmethylated) PCR product at 216bp, and the presence of the maternal (methylated) PCR product at 313 bp = complete loss of paternal allele.
Methods for detection – Methylation sensitive MLPA analysis (MS-MLPA) MS-MLPA is a semi-quantitative method for methylation profiling – variation on standard MLPA where copy number detection is combined with the use of a methylation specific restriction enzyme. (PWS/AS, BWS/RSS, tumour analysis) In MS-MLPA the reaction generates two samples: Undigested for copy number detection Digested for methylation detection If the probes are hybridised to an unmethylated site - sample is digested and will not produced a signal, however if the probes are hybridised to a methylated site a probe signal will be generated. Advantages – small quantity of DNA required, no need for parental samples, quantitative information on copy number changes, methylation status and parent of origin effect. Disadvantages – polymorphisms under probe binding sites = false –ive/+ive, Can’t differentiate between UPD and imprinting defects.
Methods for detection – High resolution melt analysis MS-HRM is a technique suitable for locus-specific assessment of DNA methylation. PCR Amplification of the region of interest from bisulfite modified DNA, using primers that amplify both methylated and unmethylated sequences PCR reactions are performed in a saturating DNA intercalating dye (LC green) A slowly increasing temperature is used which makes the double stranded DNA melt, therefore releasing the dye = recognised as fluorescence The methylated dye melts at a higher temperature (CG rich), whereas unmethylated melts at a lower temperature (AT rich due to bisulfite treatment).
Methods for detection – High resolution melt analysis This figure shows High resolution melt analysis for diagnosis of PWS/AS.
Methods for detection – Pyrosequencing Is a real time sequencing method for analysis of short to medium length DNA sequences Incorporation of a nucleotide into the template strand leads to the release of pyrophosphate which is quantified with a luciferase reaction Signal produced = proportional to the amount of pyrophosphate released Using this technology and bisulfite treated DNA it is possible to detect and quantify methylation at CpG sites by analysing the chemically induced C/T sequence differences at a number of sites.
Pyrosequencing schematic B = region being amplified - analysis of CpG sites R, S, T and U. * is a bisulfite treatment control. Pyrogram I is an AS patient - AQ values (shaded boxes) for the methylated G allele at all CpG sites are 0% - indicating the absence of the maternal (methylated allele) Pyrogram II is a healthy control – AQ values for the methylated G allele range from 31.5-33.5% which after correction for PCR amplification bias = 50% methylation = normal methylation profile. Pyrogram III is a PWS patient – AQ values for the methylated G allele at all CpG sites = 100% - indicating the absence of the paternal (unmethylated allele) In all cases the bisulfite control * shows complete conversion of the unmethylated cytosine residue (AQ value = 100%)
References Epigenetic Mechanisms in Health and Disease, S. M. Van derMaarel, 2008, Ann Rheum Dis, (Suppl III): iii97-iii100 Epigenetics and human disease Hirstet al., 2009, The international journal of biochemistry and cell biology, 41:136-146. Human Molecular Genetics 3 www.mrcholland.com P53 and deregulation of DNA methylation in cancer, Cell Science Reviews, Vol. 2 No. 3: http://www.cellscience.com/reviews7/Cancer_DNA_methylation.html Epigenetic events in Medulloblastoma development: Identification of hypermethylated genes in medulloblastoma: http://www.medscape.com/viewarticle/518166_8 Rapid high-throughput restriction analyses using the lightscanner 480 System, Krypuryet al., 2008, Biochemica No. 1: http://gene-quantification.com/krypuy-et-al-ras-hrm-2007.pdf Quantitative analysis of SNRPN gene methylation by pyrosequencing as a diagnostic test for Prader Willi syndrome and Angelman syndrome, White et al. 2006, Clinical Chemistry, 52:6, 1005-1013.