Download

1 / 22

E N D
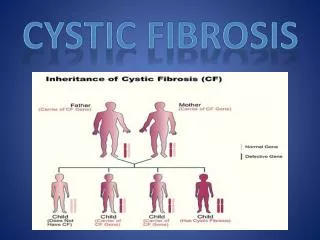
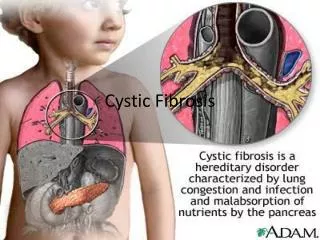
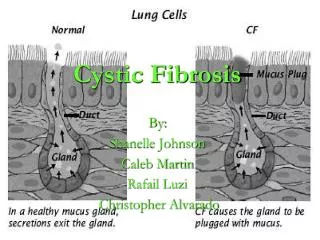
Cystic fibrosis (CF) is an autosomalrecessivegenetic disorder that affects most critically the lungs, and also the pancreas, liver, and intestine. It is characterized by abnormal transport of chloride and sodium across an epithelium, leading to thick, viscous secretions.
How common is cystic fibrosis? CF is more commonly found in the white population, the disease affects all racial groups. CF occurs in about one in every 3,500 white births, one in every 17,000 black births, and one in every 90,000 Asian births. CF is less common in most Native American tribes. However, it is being seen more often in Hispanics. It occurs equally in males and females
The CFTR gene, found at the q31.2 locus of chromosome 7 • there are over 1500 other mutations that can produce • The most common mutation, ΔF508, is a deletion (Δ signifying deletion) of three nucleotides
CFTR is an ATP-responsive chloride channel that also affects other cellular activities, such as sodium transport across the respiratory epithelium, composition of cell surface glycoprotein and antibacterial defences.
Symptoms of Cystic Fibrosis • People with CF can have a variety of symptoms, including: • very salty-tasting skin; • persistent coughing, at times with phlegm; • frequent lung infections; • wheezing or shortness of breath; • poor growth/weight gain in spite of a good appetite; and • frequent greasy, bulky stools or difficulty in bowel movements
Diagnosis • Sweat Test — If a person shows symptoms of CF or if a baby has a positive newborn screen for CF, a doctor may order a sweat test. This simple, painless test is the best way to diagnose CF. It measures the concentration of salt in a person’s sweat. A high salt level indicates CF.
Genetic testing. DNA samples from blood or saliva can be checked for specific defects on the gene responsible for cystic fibrosis.
Newborn Screening • All States screen newborns for CF using a genetic test or a blood test. The genetic test shows whether a newborn has faulty CFTR genes. The blood test shows whether a newborn's pancreas is working properly.
Testing after diagnosis • Imaging tests. Damage to your lungs or intestines can be monitored with X-rays, CT scans and MRI. • Lung function tests. These tests measure the size of your lungs, how much air you can breathe in and out, how fast you can breathe in and out, and how well your lungs deliver oxygen to your blood.
Organ function tests. Blood tests can measure the health of your pancreas and liver. Children with cystic fibrosis should be regularly tested for diabetes after age 10.
treatment • Cystic fibrosis (CF) has no cure. However, treatments have greatly improved in recent years. The goals of CF treatment include: • Preventing and controlling lung infections • Loosening and removing thick, sticky mucus from the lungs • Preventing or treating blockages in the intestines • Providing enough nutrition • Preventing dehydration (a lack of fluid in the body)