Computational Studies on Density Functional Theory for Oxidation Intermediates and Molecules Diffusion Energy
60 likes | 162 Views
Explore computations using density functional theory for oxidation intermediates, hydrogen interactions in silica, and adsorbed species on phosphide clusters. Conduct calculations for activation energy, diffusion energy, and molecular dynamics simulations to study membrane structures and permeation in inorganic-organic hybrids.

Computational Studies on Density Functional Theory for Oxidation Intermediates and Molecules Diffusion Energy
E N D
Presentation Transcript
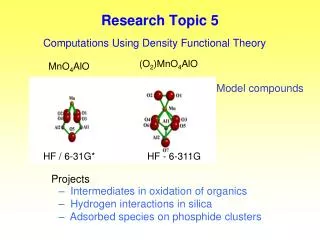
Research Topic 5 Computations Using Density Functional Theory (O2)MnO4AlO MnO4AlO Model compounds HF / 6-31G* HF - 6-311G Projects – Intermediates in oxidation of organics– Hydrogen interactions in silica– Adsorbed species on phosphide clusters
Diffusion Energy Saddle Point Membrane Diffusion Energy Calculations Model: Becke3lyp (DFT) Basis set: 6-311G(2d,p) x z y Normal to the xy plane
Glasses Silica layer Activation Energy vs. Distance to O atoms 4-membered 5-membered 6-membered 7-membered 8-membered Helium Hydrogen P. Hacarlioglu, D. Lee, G.V. Gibbs and S.T. Oyama, J. Membr. Sci. 2008,313, 278-283 .
Multicomponent Membranes 4.1 A 3.9 A 3.5 A silica germania alumina 4.3 A 4.0 A boria titania 5.2 A 5.3 A yttria zirconia E(UB+HF-LYP)=-516.292128356
Extended X-ray AbsorptionFine Structure (EXAFS) Laser Raman Spectroscopy (LRS) 984 O 819 Mo 1550 804 O O SiO Mo Mo 2 2 2000 1500 1000 500 -1 Raman Shift / cm X-ray Absorption NearEdge Spectroscopy (XANES) Ab Initio Molecular Orbital Calculations (Gaussian) Characterization of Catalysts Vibrational frequencies Bond distances Theoretical values Symmetry
Molecular Dynamics Simulations • Structure of membranes • Inorganic-organic hybrids • Permeation studies 10% Removed 30% Removed Phenyl substitution



















