Download

1 / 6
0 likes | 14 Views
Learn about key IND Data Requirements and the US FDA Submission Process to ensure safe, compliant, and successful drug development approval.

E N D
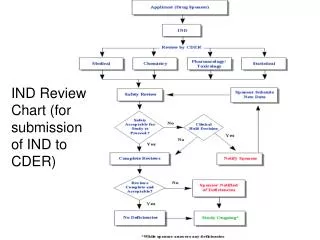
IND Data Requirements and US FDA Submission Process When it comes to introducing a new drug to the U.S. market, the Investigational New Drug (IND) application is a critical first step. Before any human clinical trials can begin, pharmaceutical sponsors must demonstrate to the U.S. Food and Drug Administration (FDA) that their investigational product is reasonably safe for initial use in humans. This process involves a well-structured set of IND data requirements covering everything from preclinical studies to chemistry, manufacturing, and control (CMC) data. For organizations planning to conduct clinical trials in the U.S., understanding the IND Data Requirements and the US FDA submission process is key to avoiding costly delays and compliance setbacks. What Are the Key Components of IND Data Requirements? The key components of IND Data Requirements include preclinical pharmacology and toxicology data, chemistry, manufacturing, and controls (CMC) information, and detailed clinical study protocols. These elements collectively demonstrate drug safety, quality, and scientific justification for initiating human clinical trials.
Section Description Purpose / FDA Expectation 1. Introductory Statement and General Investigational Plan Provides an overview of the investigational drug, objectives, and proposed clinical studies. Helps FDA understand the overall research intent and therapeutic area. 2. Investigator’s Brochure (IB) Summarizes preclinical and clinical information on the investigational drug. Assists investigators in understanding the rationale and potential risks/benefits. 3. Clinical Protocols Details the design, methodology, and objectives of the proposed human studies. Ensures ethical and scientific integrity of the trial. 4. Chemistry, Manufacturing, and Controls (CMC) Describes drug substance, formulation, manufacturing process, stability, and quality control. Ensures drug consistency, purity, and identity. 5. Pharmacology and Toxicology Data Includes animal study results on safety, pharmacokinetics, and toxicity. Provides evidence that the product is safe to test in humans. 6. Previous Human Experience Summarizes prior clinical data, if available. Helps FDA assess cumulative safety data. 7. Additional Supporting Information May include environmental assessments, investigator qualifications, and IRB approvals. Confirms overall compliance with FDA regulations. This structure allows the FDA to systematically assess both scientific validity and patient safety before authorizing human exposure. How Does the US FDA Submission Process for IND Work?
Once all required data is collected, the sponsor prepares the IND submission in the Common Technical Document (CTD) format. The FDA’s Center for Drug Evaluation and Research (CDER) or Center for Biologics Evaluation and Research (CBER) reviews the application depending on the product type. Here’s a step-by-step overview of the US FDA submission process: 1. Pre-IND Meeting Before filing the IND, sponsors can request a Pre-IND meeting with the FDA. This is an excellent opportunity to discuss preclinical findings, manufacturing details, and proposed clinical protocols. Early communication often prevents rework and clarifies data expectations. 2. IND Submission The completed IND is electronically submitted through the FDA’s Electronic Submissions Gateway (ESG). The submission should comply with eCTD (electronic Common Technical Document) format standards for easy review and traceability. 3. FDA Review and 30-Day Safety Assessment Once submitted, the FDA has 30 days to review the IND and ensure that participants in the proposed clinical trials are not exposed to unreasonable risk. During this period, the agency evaluates: ● Adequacy of preclinical safety data ● Quality and consistency of CMC data ● Scientific soundness of clinical protocols ● Qualifications of investigators If no clinical hold is issued within 30 days, the sponsor may proceed with the clinical trial. 4. Clinical Hold or Clearance If FDA identifies any safety concerns or data gaps, it may place the IND on clinical hold. The sponsor must then address these deficiencies before proceeding. Once the issues are resolved and FDA clears the application, human trials can commence under the approved protocol. 5. Ongoing IND Maintenance Throughout the trial, sponsors must maintain compliance through:
● Amendments: For protocol changes or additional studies ● Safety Reports: For serious adverse events ● Annual Reports: Summarizing progress and findings This ongoing interaction ensures continuous oversight until the trial concludes or a New Drug Application (NDA) is filed. Why Are IND Data Requirements Crucial for FDA Success? The IND Data Requirements are not mere formalities they are designed to safeguard participants and ensure scientific credibility. Missing or incomplete data can lead to IND rejections or holds, significantly delaying development timelines. Here are some of the most common pitfalls sponsors should avoid: ● Submitting incomplete toxicology reports or missing dose justification. ● Insufficient CMC data on stability, sterility, or impurity profiles. ● Vague or scientifically weak clinical protocols. ● Failure to ensure investigator qualifications or IRB approvals. A well-prepared IND not only expedites FDA clearance but also builds sponsor credibility for later NDA/BLA submissions. Best Practices for a Successful IND Submission To streamline your IND process and enhance the chance of FDA acceptance, consider these best practices: 1. Engage Early with FDA: Seek pre-IND guidance to align on data requirements. 2. Use the eCTD Format: Follow FDA’s Module 1–5 structure for consistency. 3. Ensure Data Traceability: Maintain version control and cross-reference data. 4. Focus on CMC Quality: Poor manufacturing data is a frequent cause of delays. 5. Document Every Decision: Maintain an audit-ready trail for FDA inspections.
Sponsors partnering with experienced Clinical Research Organizations (CROs) benefit from regulatory expertise, quality documentation, and faster turnaround times. Final Takeaways 1. What is the purpose of the IND application? The IND application seeks FDA authorization to begin clinical trials in humans. It demonstrates that the investigational drug is reasonably safe based on preclinical and manufacturing data. 2. How long does the FDA take to review an IND? The FDA typically completes its review within 30 calendar days. If no hold is issued, the sponsor can proceed with clinical trials. 3. Can an IND be amended after submission? Yes. Sponsors may submit protocol amendments to modify study design, add investigators, or update clinical information. 4. What happens if the IND is placed on hold? The sponsor must address FDA’s concerns by providing additional data or clarifications. The trial cannot start until the hold is lifted. 5. Who regulates IND submissions for biologics? For biologics, the Center for Biologics Evaluation and Research (CBER) manages the IND review, while CDER handles chemical drugs. Conclusion Understanding the IND Data Requirements and the US FDA Submission Process is crucial for a successful drug development journey. A well-documented and compliant IND not only protects trial participants but also paves the way for efficient clinical progression and eventual market approval.
With meticulous data preparation, regulatory alignment, and expert support, sponsors can confidently navigate the complex FDA landscape and move closer to bringing safe, effective therapies to patients in need.