Download

1 / 95
1.02k likes | 2.28k Views
Pharmacokinetics. Drug Effectiveness. Dose-response (DR) curve : Depicts the relation between drug dose and magnitude of drug effect Drugs can have more than one effect Drugs vary in effectiveness Different sites of action Different affinities for receptors

E N D
Drug Effectiveness • Dose-response (DR) curve: Depicts the relation between drug dose and magnitude of drug effect • Drugs can have more than one effect • Drugs vary in effectiveness • Different sites of action • Different affinities for receptors • The effectiveness of a drug is considered relative to its safety (therapeutic index)
Therapeutic Index This is a figure of two different dose response curves. You can obtain a different dose response curve for any system that the drug effects. When you vary the drug, this is the Independent variable, what you are measuring is the % of individuals responding to the drug. Here we see the drugs effects on hypnosis and death. Notice that the effective dose for 50 % of the people is 100 mg and if you double the dose to 200 mg then 1 % of your subjects die. Thus, if you want to use this drug to hypnotize 99 % of your subjects, in the process you will kill 2-3 % of your subjects.
Drug Safety and Effectiveness • Not all people respond to a similar dose of a drug in the exact same manner, this variability is based upon individual differences and is associated with toxicity. This variability is thought to be caused by: • Pharmacokinetic factors contribute to differing concentrations of the drug at the target area. • Pharmacodynamic factors contribute to differing physiological responses to the same drug concentration. • Unusual, idiosyncratic, genetically determined or allergic, immunologically sensitized responses.
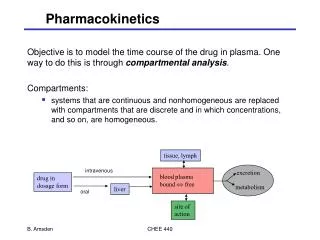
Pharmacokinetics • Drug molecules interact with target sites to effect the nervous system • The drug must be absorbed into the bloodstream and then carried to the target site(s) • Pharmacokinetics is the study of drug absorption, distribution within body, and drug elimination over time. • Absorption depends on the route of administration • Drug distribution depends on how soluble the drug molecule is in fat (to pass through membranes) and on the extent to which the drug binds to blood proteins (albumin) • Drug elimination is accomplished by excretion into urine and/or by inactivation by enzymes in the liver
DISPOSITION OF DRUGS The disposition of chemicals entering the body (from C.D. Klaassen, Casarett and Doull’s Toxicology, 5th ed., New York: McGraw-Hill, 1996).
Routes of Administration • Routes of Administration:Orally: • Rectally: • Inhalation: Absorption through mucous membranes: • Topical: • Parenterally: • Intravenous: • Intramuscular: • Subcutaneous:
Drug Delivery Systems • Tablets • Injections (Syringe) • Cigarettes • Beverages • Patches • Suppositories • Candy • Gum • Implants • Gas • Creams • Others? • Stamps • Bandana
Membranes • Types of Membranes: • Cell Membranes: This barrier is permeable to many drug molecules but not to others, depending on their lipid solubility. Small pores, 8 angstroms, permit small molecules such as alcohol and water to pass through. • Walls of Capillaries: Pores between the cells are larger than most drug molecules, allowing them to pass freely, without lipid solubility being a factor. • Blood/Brain Barrier: This barrier provides a protective environment for the brain. Speed of transport across this barrier is limited by the lipid solubility of the psychoactive molecule. • Placental Barrier: This barrier separates two distinct human beings but is very permeable to lipid soluble drugs.
Drug Distribution • Dependent upon its route of administration and target area, every drug has to be absorbed, by diffusion, through a variety of bodily tissue. • Tissue is composed of cells which are encompassed within membranes, consisting of 3 layers, 2 layers of water-soluble complex lipid molecules (phospholipid) and a layer of liquid lipid, sandwiched within these layers. Suspended within the layers are large proteins, with some, such as receptors, transversing all 3 layers. • The permeability of a cell membrane, for a specific drug, depends on a ratio of its water to lipid solubility. Within the body, drugs may exist as a mixture of two interchangeable forms, either water (ionized-charged) or lipid (non-ionized) soluble. The concentration of two forms depends on characteristics of the drug molecule (pKa, pH at which 50% of the drug is ionized) and the pH of fluid in which it is dissolved. • In water soluble form, drugs cannot pass through lipid membranes, but to reach their target area, they must permeate a variety of types of membranes.
Pharmacokinetics vs Pharmacodynamics…concept • Fluoxetine increases plasma concentrations of amitriptyline. This is a pharmacokinetic drug interaction. • Fluoxetine inhibits the metabolism of amitriptyline and increases the plasmaconcentration of amitriptytline.
Pharmacokinetics vs Pharmacodynamics…concept • If fluoxetine is given with tramadol serotonin syndrom can result. This is a pharmacodynamic drug interaction. • Fluoxetine and tramadol both increase availability of serotonin leading to the possibility of “serotonin overload” This happens without a change in the concentration of either drug.
Basic Parameters • In the next few slides the basic concepts and paramaters will be described and explained. • In pharmacokinetics the body is represented as a single or multiple compartments in to which the drug is distributed. • Some of the parameters are therefore a little abstract as we know the body is much more complicated !
Volume of Distribution, Clearance and Elimination Rate Constant V Volume 100 L Clearance 10 L/hr
Volume of Distribution, Clearance and Elimination Rate Constant V Volume 100 L (Vi) V2 Cardiac and Skeletal Muscle Clearance 10 L/hr
Volume 100 L (Vi) V2 Cardiac and Skeletal Muscle V Clearance 10 L/hr Volume of Distribution = Dose_______ Plasma Concentration
Volume 100 L (Vi) V2 Cardiac and Skeletal Muscle V Clearance 10 L/hr Clearance = Volume of blood cleared of drug per unit time
Volume 100 L (Vi) V2 Cardiac and Skeletal Muscle V Clearance 10 L/hr Clearance = 10 L/hr Volume of Distribution = 100 L What is the Elimination Rate Constant (k) ?
CL = kV k = 10 Lhr -1 = 0.1 hr -1 100 L 10 % of the “Volume” is cleared (of drug) per hour k = Fraction of drug in the body removed per hour
CL = kV If V increases then k must decrease as CL is constant
Important Concepts • VD is a theoretical Volume and determines the loading dose. • Clearance is a constant and determines the maintenance dose. • CL = kVD. • CL and VD are independent variables. • k is a dependent variable.
Volume of Distribution (Vd) Apparent volume of distribution is the theoretical volume that would have to be available for drug to disperse in if the concentration everywhere in the body were the same as that in the plasma or serum, the place where drug concentration sampling generally occurs.
Volume of Distribution • An abstract concept • Gives information on HOW the drug is distributed in the body • Used to calculate a loading dose
Loading Dose Dose = Cp(Target) x Vd
Question • What is the loading dose required for drug A if; • Target concentration is 10 mg/L • Vd is 0.75 L/kg • Patients weight is 75 kg • Answer is on the next slide
Answer: Loading Dose of Drug A • Dose = Target Concentration x VD • Vd = 0.75 L/kg x 75 kg = 56.25 L • Target Conc. = 10 mg/L • Dose = 10 mg/L x 56.25 L • = 565 mg • This would probably be rounded to 560 or even 500 mg.
Clearance (CL) • Ability of organs of elimination (e.g. kidney, liver) to “clear” drug from the bloodstream. • Volume of fluid which is completely cleared of drug per unit time. • Units are in L/hr or L/hr/kg • Pharmacokinetic term used in determination of maintenance doses.
Clearance • Volume of blood in a defined region of the body that is cleared of a drug in a unit time. • Clearance is a more useful concept in reality than t 1/2 or kel since it takes into account blood flow rate. • Clearance varies with body weight. • Also varies with degree of protein binding.
Clearance • Rate of elimination = kel D, • Remembering that C = D/Vd • And therefore D= C Vd • Rate of elimination = kel C Vd • Rate of elimination for whole body = CLT C Combining the two, CLT C = kel C Vd and simplifying gives: CLT = kel Vd
Maintenance DoseCalculation • Maintenance Dose = CL x CpSSav • CpSSav is the target average steady state drug concentration • The units of CL are in L/hr or L/hr/kg • Maintenance dose will be in mg/hr so for total daily dose will need multiplying by 24
Question • What maintenance dose is required for drug A if; • Target average SS concentration is 10 mg/L • CL of drug A is 0.015 L/kg/hr • Patient weighs 75 kg • Answer on next slide.
Answer • Maintenance Dose = CL x CpSSav • CL = 0.015 L/hr/kg x 75 = 1.125 L/hr • Dose = 1.125 L/hr x 10 mg/L = 11.25 mg/hr • So will need 11.25 x 24 mg per day= 270 mg
Half-Life and k • Half-life is the time taken for the drug concentration to fall to half its original value • The elimination rate constant (k) is the fraction of drug in the body which is removed per unit time.
Half-Life • C = Co e - kt • C/Co = 0.50 for half of the original amount • 0.50 = e – k t • ln 0.50 = -k t ½ • -0.693 = -k t ½ • t 1/2 = 0.693 / k
Use of t ½ and kel data • If drug has short duration of action, design drug with larger t ½ and smaller kel • If drug too toxic, design drug with smaller t ½ and larger kel
Drug Concentration C1 Time Exponential decay dC/dt C = -k.C C2
Log Concn. Time Time to eliminate ~ 4 t1/2 C0 C0/2 t1/2 t1/2 t1/2
Cp2 = Cp1.e-kt t1/2 = 0.693/k Integrating: Logarithmic transform: lnC2= lnC1 - kt logC2 = logC1 - kt/2.303 Elimination Half-Life: t1/2 = ln2/k
Steady-State • Steady-state occurs after a drug has been given for approximately five elimination half-lives. • At steady-state the rate of drug administration equals the rate of elimination and plasma concentration - time curves found after each dose should be approximately superimposable.
Accumulation to Steady State 100 mg given every half-life … 200 194 187.5 175 150 100 … 97 100 94 87.5 75 50
What is Steady State (SS) ?Why is it important ? • Rate in = Rate Out • Reached in 4 – 5 half-lives (linear kinetics) • Important when interpreting drug concentrations in time-dependent manner or assessing clinical response