Electronic Structure Methods-Part 2
metode post HF

Electronic Structure Methods-Part 2
E N D
Presentation Transcript
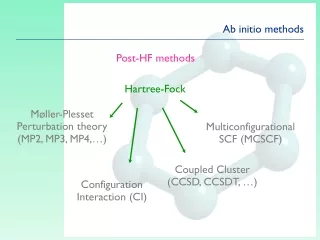
Ab initio methods Post-HF methods Hartree-Fock Møller-Plesset Perturbation theory (MP2, MP3, MP4,…) Multiconfigurational SCF (MCSCF) Coupled Cluster (CCSD, CCSDT, …) Configuration Interaction (CI)
Ab initio methods 2. Configuration Interaction (CI)
Configuration Interaction The Hartree-Fock method determines the best one-determinant wavefunction (for a given basis set) To overcome the weaknesses of the Hartree-Fock method (in particular, the lack of electron correlation), additional Slater Determinants can be added to the wavefunction Configuration Interaction: the additional determinants are obtained by single, double, triple, etc. excitations from the optimised HF determinant (yielding excited Slater Determinants)
Configuration Interaction Excited Slater determinants N electrons, M basis functions: The Hartree-Fock method yields N occupied MOs (in the Slater Determinant) and M – N unoccupied (virtual) MOs Occupied MOs can be replaced by unoccupied MOs, generating excited Slater Determinants
Configuration Interaction HF configuration single excitation double excitation
Configuration Interaction Hartree-Fock determinant Hartree-Fock determinant, with one MO swapped with a virtual orbital (“single excitation”) two MOs swapped with virtual orbitals (“double excitation”) CI expansion coefficients
Configuration Interaction The MOs used for building the excited Slater Determinants are taken directly from the Hartree-Fock calculations. The MO coefficients are not reoptimised! The Configuration Interaction method finds the optimal CI expansion coefficients a0, aS, aD, … This is done using the variation principle (find those coefficients that yield the wavefunction with the lowest energy)
Configuration Interaction Truncated CI methods Full CI (FCI): all possible excited Slater Determinants are included => very expensive; not feasible for any but the smallest systems! CISD (or SDCI): including all single and double excited Slater determinants CISDT, CISDTQ, etc.
Configuration Interaction Hartree-Fock (HF): one Slater Determinant MO coefficients cmi optimised using variation principle Configuration Interaction (CI): linear combination of Slater Determinants CI coefficients ai optimised using variation principle
Ab initio methods 3. Multi-configuration Self Consistent Field (MCSCF)
Multi-configuration Self Consistent Field Multi-configuration Self Consistent Field (MCSCF) MCSCF is a CI, where not only the CI expansion coefficients ai, but also the MOs in the determinants (the MO coefficients cmi) are optimised by the variation principle MCSCF methods are often used for cases where HF does not give a qualitatively correct description
+ • • - Multi-configuration Self Consistent Field Consider for example a molecule with two non-equivalent resonance states: requires two singly occupied MOs – cannot be described by an HF wavefunction
Multi-configuration Self Consistent Field Special case:Complete Active Space SCF (CASSCF) The MOs are divided into active and inactive spaces Within the active MOs, a full CI is performed (all possible distributions of the electrons over the active orbitals) Active MOs: usually some of the highest occupied and some of the lowest unoccupied
Multi-configuration Self Consistent Field inactive active orbitals CAS inactive (kept unchanged from HF orbital) HF configuration In the CASSCF wavefunction all configurations are included that can be generated by distributing the two electrons over the three orbitals in the CAS
Multi-configuration Self Consistent Field MCSCF is used in cases where HF does not give a correct reference wavefunction, for example for molecular ground states that are almost degenerate with low-lying excited states, for bond breaking situations, or for describing excited states MCSCF recovers some electron correlation, but mostly “static electron correlation” (resulting from the additional flexibility required to qualitatively describe the system, and not so much “dynamic electron correlation” (energy lowering caused by correlating the motions of the electrons)
Ab initio methods 4. Multi-reference Configuration Interaction (MRCI)
Multi-Reference Configuration Interaction Multi-Reference Configuration Interaction (MRCI) CI: inclusion of excited Slater determinants created by exciting electrons from a single determinant (the HF determinant) MRCI: MCSCF wavefunction as the reference. MRCI is very computationally demanding
Ab initio methods 5. Møller-Plesset (MP) perturbation theory
Møller-Plesset perturbation theory Møller-Plesset (MP) perturbation theory Divide Hamiltonian into two parts: H = H0 + lV Sum of one-electron Fock operators small perturbation (electron correlation)
Møller-Plesset perturbation theory Hartree-Fock energy E(2): first correction of the HF energy Always negative! MP2 = second-order Møller-Plesset perturbation theory Higher-order terms: MP3, MP4, MP5, … Question: Does the MP series converge?
Møller-Plesset perturbation theory Does the MP series converge? Not always!!! “Surprising cases of divergent behaviour in Møller-Plesset perturbation theory” J. Olsen, O. Christiansen, H. Koch, P. Jørgensen J. Chem. Phys. 105, 5082 (1996) “The results thus questions the usefulness of higher-order perturbation calculations as a vehicle for obtaining arbitrary accuracy of quantum chemical calculations”
Ab initio methods 6. Coupled Cluster (CC) Theory
Coupled Cluster Theory Perturbation theory: adds all types of corrections (S, D, T, Q, …) to the reference wavefunction to a given order (first, second, …) Coupled cluster theory: adds all corrections of a certain type to infinite order The coupled cluster wavefunction: the HF wavefunction (Taylor expansion of eT)
Coupled Cluster Theory Ti: generates all ith excited Slater determinants When all operators up to Tn are included => full CI => not feasible for any but the smallest systems! cluster operator must be truncated at some excitation level
Coupled Cluster Theory Truncated CC methods T=T2 CCD T=T1+T2 CCSD very expensive! T=T1+T2+T3 CCSDT triples from perturbation theory CCSD(T) CCSD(T) generally a very accurate method (unless the molecular system contains considerable multi-reference character)
MP2 MP4 CCSD MP3 CCSD(T) MRCI MRCI Time / s HF MP4 CCSD(T) MP2 MP3 HF CCSD Exptl. MP2 MP3 MP4 examples Binding energy of N2 122.2 HF MP2 239.6 MP3 215.2 MP4 231.5 De / kcal mol-1 CCSD 217.2 CCSD(T) 226.4 MRCI 227.2 AK Wilson, T van Mourik, TH Dunning, Jr., THEOCHEM 368, 339 (1996) Exptl. 228.4
Electronic structure methods Density functional theory (DFT)
Density Functional Theory Density Functional Theory (DFT) DFT describes a molecular system directly via its density, without first finding the wavefunction DFT models electron correlation via functionals of the electron density (Functional: “A function of a function”) Functional in DFT: function of the electron density Function of the coordinates of the molecular system
Density Functional Theory Density functionals Hohenberg-Kohn theorem:there exists a functional that relates the ground state energy and electron density However: the form of this functional is unknown => it is generally approximated by model functionals B3LYP, PBE, HCTH, … M05-2X, PWB6K, OLYP, …
B3LYP B3LYP DFT examples Binding energy of N2 MP2 MRCI MP4 CCSD MP3 De / kcal mol-1 MP4 CCSD(T) Time / s CCSD(T) MRCI MP2 MP3 HF CCSD HF
CCSD(T) Methodological accuracy Many density functionals do not describe “dispersion” He2 potential energy curve DE B3LYP MP2 R
DFT examples Tyr-Gly conformers f = 80º f = 180º f = 280º
DFT examples B3LYP M06-2X DE / kJ mol-1 CCSD(T)
Methodological accuracy HF does not include electron correlation DFT is computationally efficient. However, most density functionals do not include dispersion MP2 does include dispersion, but is more computationally expensive (and in some cases overestimates dispersion) CCSD(T) usually gives very good results, but is very expensive