Download

1 / 3
30 likes | 48 Views
RNA sequencing, also called whole transcriptome shotgun sequencing, uses next-generation sequencing (NGS) to reveal the presence and quantity of RNA in a biological sample. Read more

E N D
RNA Seq: Modern Tool For Transcriptome Analysis And Gene Expression In Eukaryotes RNA sequencing (RNA seq), also called whole transcriptome shotgun sequencing, uses next- generation sequencing (NGS) to reveal the presence and quantity of RNA in a biological sample at a given time. The correct identification of differentially expressed genes (DEGs) between specific conditions is a key in the understanding phenotypic variation. High-throughput transcriptome sequencing (RNA seq) has become the main option for these studies. Development of high-throughput next-generation sequencing (NGS) has revolutionized transcriptomics by enabling RNA seq analysis through the sequencing of complementary DNA (cDNA) (Wang et al. 2009). This method, termed RNA sequencing or RNA seq, has distinct advantages over previous approaches and has revolutionized our understanding of the complex and dynamic nature of the transcriptome. RNA seq provides a more detailed and quantitative view of gene expression, alternative splicing, and allele-specific expression. Recent advances in the RNA seq workflow, from sample preparation to sequencing platforms to bioinformatic RNA seq data analysis, has enabled deep profiling of the transcriptome (also termed as transcriptome analysis) and the opportunity to elucidate different physiological and pathological conditions. Why to do RNA Sequencing? 1) With RNA Seq you can interrogate more than just differential gene expression. Although there are microarrays available for exon-level and microRNA analysis, most users are still interested in basic, probably 3’ biased, differential gene expression. 2) With RNA Seq you can look at coding and non-coding RNA, at splicing and allele specific expression, and possibly soon at full-length cDNA sequences, eliminating the need to infer or assemble isoforms. Since RNA seq does not use probes or primers, the data is less biased. 3) RNA Seq can reveal the precise location of transcription boundaries, to a single-base resolution. Furthermore, 30-bp short reads from RNA Seq give information about how two exons are connected, whereas longer reads or pair-end short reads should reveal connectivity between multiple exons. 4) A second advantage of RNA Seq relative to DNA microarrays is that it has very low, if any, background signal because DNA sequences can been unambiguously mapped to unique regions of the genome. RNA Seq does not have an upper limit for quantification, which correlates with the number of sequences obtained. Clinical significance for RNA Seq RNA seq is now the preferred method of transcriptome profiling and transcriptome analysis, favored over microarray analysis because of its higher sensitivity, broader dynamic range, capacity for transcript discovery, and lack of requirement for pre-existing sequence knowledge.
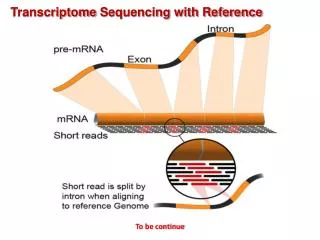
RNA Seq Data Generation A typical RNA seq experimental workflow involves the isolation of RNA from samples of interest, generation of sequencing libraries, use of a high-throughput sequencer to produce hundreds of millions of short paired-end reads, alignment of reads against a reference genome or transcriptome, and downstream analysis for expression estimation, differential expression, transcript isoform discovery, and other applications.
Case studies performed using RNA Seq for screening rare disorders 1. Genetic diagnosis of Mendelian disorders via RNA sequencing: Laura S. Kremer et-al Nature Communications volume 8, Article number: 15824 (2017) RNA Seq analysis was performed on 105 fibroblast cell lines from patients with a suspected mitochondrial disease including 48 patients for which WES based variant prioritization did not yield a genetic diagnosis. After discarding lowly expressed genes, RNA seq identified 12,680 transcribed genes (at least 10 reads in 5% of all samples). Researchers found a median of one aberrantly expressed gene, five aberrant splicing events and six mono-allelically expressed rare variants in patient-derived fibroblasts and establish disease-causing roles for each kind. Private exons often arise from cryptic splice sites providing an important clue for variant prioritization. One such event is found in the complex I assembly factor TIMMDC1 establishing a novel disease-associated gene. 2. The diverse applications of RNA-seq for functional genomic studies in Aspergillus fumigatus Ann N Y Acad Sci. 2012 Dec; 1273(1): 25–34. PMID: 23230834 In this set of experiment performed by Ann N Y et al, they used RNA-seq to study the transcriptome of Aspergillus fumigatus, a deadly human fungal pathogen. Analysis of the RNA-seq data indicates that there are likely tens of unannotated and hundreds of novel genes in the A. fumigates transcriptome, mostly encoding for small proteins. Inspection of transcriptome-wide variation between two isolates reveals thousands of single nucleotide polymorphisms. Finally, comparison of the transcriptome profiles of one isolate in two different growth conditions identified thousands of differentially-expressed genes. Analysis of RNA seq data produced from a single experiment in A. fumigatus has uncovered tens of putative unannotated and hundreds of novel small genes, thousands of SNPs, and hundreds of candidates for downstream functional experiments to identify the molecular basis of colony growth and its potential role in the establishment of some forms of aspergillosis.