Download
1 / 29
290 likes | 392 Views
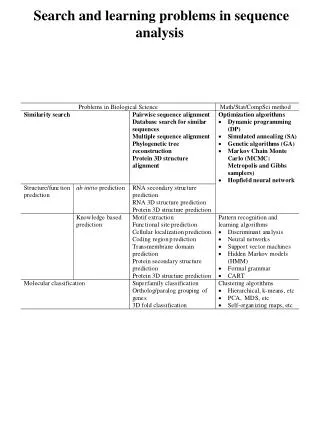
Search and learning problems in sequence analysis. Multiple sequence alignment Dado un conjunto de secuencias encontrar la subsecuencia común más larga entre las cadenas problema. Muestra qué partes de estas cadenas están relacionadas con una otra. Sequence reconstruction

E N D
Multiple sequence alignment Dado un conjunto de secuencias encontrar la subsecuencia común más larga entre las cadenas problema. Muestra qué partes de estas cadenas están relacionadas con una otra.
Sequence reconstruction Una secuencia de bases de una extensa región será determinada debido a que la región ha sido fragmentada, se han secuenciado y reconstruido los fragmentos. Ejemplo 1. -Secuenciado por Hibridación. La complementariedad de la cadena de DNA (bases) permite que se unan oligonucleótidos (fragmentos muy pequeños de DNA) a una secuencia más larga. -Oligos del mismo tamaño hibridan con una secuencia dada. Las regiones de solapamiento en las hibridaciones detectadas se usarán para reconstruir la secuencia original. Un caso práctico serría que la secuencia ATCCGC puede ser reconstruida por el conjunto; ATC, TCC, CCG, CGC
Sequence reconstruction Ejemplo 2. -Reconstruir el orden de los fragmentos en la secuencia original a partir de los tamaños de solapamiento entre cada par de fragmentos de la secuencia. -El mapeo se da en varios niveles: secuencia génica, proteica y cromosómica.
Closest substring and Consensus patterns Las aplicaciones incluyen la localización de lugares de unión (binding sites) y la determinación de regiones conservadas de secuencias no alineadas. Aplicaciones en biología. -Identificación de sitios diana (target), secuencias de reconocimiento para fármacos. -Diseño de pruebas genéticas. -Diseño de encebadores (primers) para realizar la reacción de PCR.
A comparison of the homology search and the motif search for functional interpretation of sequence information. Homology Search Motif Search New sequence New sequence Knowledge acquisition Motif library (Empirical rules) Sequence database (Primary data) Retrieval Similar sequence Inference Expert knowledge Expert knowledge Sequence interpretation Sequence interpretation
Multiple alignment and consensus discovery El número de secuencias que pueden ser examinadas en una misma vez es a menudo limitado, 6. Requerimiento de tiempo O(nk) para los mejores algoritmos conocidos para estos análisis, donde: k es el número de secuencias. n es el número máximo de símbolos en cualquiera de las secuencias. Estos requerimientos parecen ser inherentes en el paradigma de la programción dinámica.
Como se afrontan los problemas difíciles en el campo de la genómica y proteómica Aproximación polinómica. Métodos heurísticos. • La naturaleza de los problemas experimentales produce datos no precisos. • Los objetivos que se buscan por el análisis computacional no necesita resultados óptimos.
Métodos heurísticos Utilizan principios matemáticos, sobre todo el análisis probabilístico para encontrar resultados cercanos al óptimo. • Alta implantación en el software comercial. • Ejemplos: • Comparació de sequencies : Blasta, Fasta, etc. • Anàlisis de expresión génica: Inducir genétic networks. • Efecto de drogas, etc. • Sustenta la cercanía a la solución optima como el nº de pasos para llegar a esta. • Intersección con el estudio de la complejidad paramétrica: • Parametrizar el numero de pasos para alcanzar la solución óptima:
Aproximación polinomial Buscan resultados aproximados al óptimo. • Intersección con el estudio de la complejidad paramétrica: • Parametrizar el factor que determina la bondad de la aproximación: Un problema tiene una aproximación polinomial eficiente si existe el problema parametrizado donde el parametro es el factor de aproximación a la solución óptima, y este problema es FPT. Gracias a este análisis de la complejidad: podemos determinar los limites de la aproximación polinómica Derivar directamente algoritmos aproximados a partir de un FPT.(estudio incipiente)
Como se afrontan los problemas dificiles en el campo de la genómica y proteómica • 2 problemas: • Analisis de sequencias : • Nos interesarán alfabetos fijos. • = bases nitrogenadas, ||=4. DNA. • = aminoacidos, ||=20.Proteinas. • Para la mayoría de problemas no tiene sentido un nº de sequencias muy grande. • Este será uno de los parámetros a estudiar. • El tamaño puede variar mucho de un EST a genomas completos. • Tratar la longitud como parametro será menos habitual. • Dendogramas.
Longest common subsequence El problema LCS K-unrestricted es NP-complete. Los mejores algoritmos conocidos requieren O(nk) y usan programación dinámica.
Longest common subsequence Complejidad si tratamos LCS de forma parametrizada. Algunos de estos problemas se vuelven FTP cuando n y son constantes.
Longest common subsequence Sentido biológico del tratamiento: K-parametrized: -no se suele trabajar con más de 6 secuencias. Alfabeto fijo: -Las cadenas de DNA y proteinas tienen un tamaño del alfabeto de 4 y 20, respectivamente. -LCS-5 especificación del LCS-4 con constante.
Multiple sequence alignment LCS no es solo una medida de consenso, sino también una guía para mostrar las regiones de cadenas relacionadas. Uno de los enfoques para tratar los k-sequences alignment es computar los pares de alineamientos. Se parte de grafos de alineamiento de pares (V, E, <), V: los caracteres de las secuencias E: los alineamientos entre los pares de secuencias a nivel de carácter. <: relación de sucesión entre los caracteres de una secuencia.
Multiple sequence alignment -Problema solventable en tiempoO(nk) mediante programación dinámica. -Los parámetros que resultan de interés biológico: k-parametrizada. -parametrizada. -Principio seguido por las aproximaciones polinomiales y métodos heurísticos.
Sequence reconstruction -SHORTEST COMMON SUPERSTRING (SCS). • -Problema NP-completo cuando >= 2. • -trabaja con multitud de fragmentos cortos(EST). • k-parametrización no resulta interesante • El máximo de fragmentos que se sobreponen si resulta interesante. • -Son útiles Algoritmos de reconstrucción polinomiales cuando el número de ocurrencias de cada x X es conocida y hay un único solapamiento en X. • -Sin embargo, en distribuciones de entrada reales, estas • condiciones no acaban de cumplirse.
Shortest SBH reconstruction (SBH) Shortest SBH reconstruction with addition (SBH-ADD) and reduction (SBH-DEL) La complejidad de estos problemas no es conocida. Los parámetros con interés biológico son: k: longitud de pares de cadenas. m: rango de errores. (para estudiar hipótesis)
Mapping G: (V, E) V: EST. E: solapamiento entre ESTs (overlap). -Si la entrada no contiene errores el problema puede ser resuelto en tiempo polinomial. Si no es así: -GI-ADD y GI-DEL son NP-completos. -La complejidad GI-DEL/ADD es todavía desconocida. -Como en SBH, tienen sentido biológico la k-parametrización para el gradual incremento de hipótesis de error.
Reducción por codificación • Para reducir problemas de complejidad conocida a problemas vinculados con la comparación de cadenas, un procedimiento habitual es codificar el primer problema • La codificación puede ser : • A nivel de palabra.: • Un elemento de ’ representa un elemento de L • Una serie de elementos de ’ representan un elemento de L. • A nivel de oración. • Definen una estructura. • Se apoyan en: • Longitudes fijas de componentes de la oración. • Repeticiones de elementos de ’ que no participan en la codificación de ningún elemento de L.
Closest substring • Closest substring es NP-complete • Una especialización es el Closest string: • Tanto s como s’ S son de igual longitud. (|s| = |s’|,s’ S) • Es también NP-complete. • Muy ligado al problema de aliniamiento múltiple. • Ambos problemas permiten aproximaciones polinomiales.
Closest substring • Sentido biológico de usar la compejidad paramétrica: • Parametrizar k: nº de cadenas. • Parametrizar d: distancia de la cadena s. • Ambas son pequeñas a la práctica. • Closest Substring resulta W[1]-hard para un alfabeto no definido. • El interés biológico reside en su complejidad con alfabetos fijos: • = bases nitrogenadas, ||=4. • = aminoacidos, ||=20.
Closest Substring : Unbounded alphabet Reducción a partir de un problema de grafos a un problema de aproximate string matching. Reducción por clique a closest substring. Procedimeinto: Obtendremos las parejas de k sequencias, apartir de un G con k-clique y m aristas. Existirá una cadena s de tamaño L. Se crea un conjunto Sc de pares de k secuencias donde cada cSc codifica las aristas de G.
Closest Substring : Unbounded alphabet Se crea un conjunto Sc de pares de k secuencias donde cada cSc codifica las aristas de G. La codificación de cada arista consiste en situar el codigo de los vértices de esta en una secuencia de elementos no codificadores de tamaño k. La posición de los códigos de los vértices será la corresponciente a las secuencias del par ci,j.
Closest Substring : Unbounded alphabet s será la codificación de los vertices del clique. • La subcadena de c cercana a s será la codificación de la • arista con la que coincide en la codificación de sus vértices. • La codificación de la arista tendrá una distancia k-2. • El resto tendrán distancia k. • Hemos codificado G creando un problema de closest substring • a partir de un G k-clique. • Encontrar la s cercana a los substrings del Sc, equivaldría a verificar que G es un k-clique. • Verificar que G es un k-clique es W[1]-> closest substring también será W[1]. • Solo existirá s próximo a s’Cs ssi G es k-clique. • Ssi (x’,k’) L’ entonces (x,k) L.
LCS-4 complexity Reduccion de LCS-1 a LCS-4. LCS-4 es W[t]-hard Mientras que LCS-1 parametriza k, LCS-4 parametriza K y ||. La clave está en : Codificar el alfabeto flexible de LCS-1 con un alfabeto de tamaño fijo. Construir las secuencias de tal forma que se cumpla que la subcadena se encuentra tanto para el primer como para el segundo problema.





















