Download
1 / 17
180 likes | 348 Views
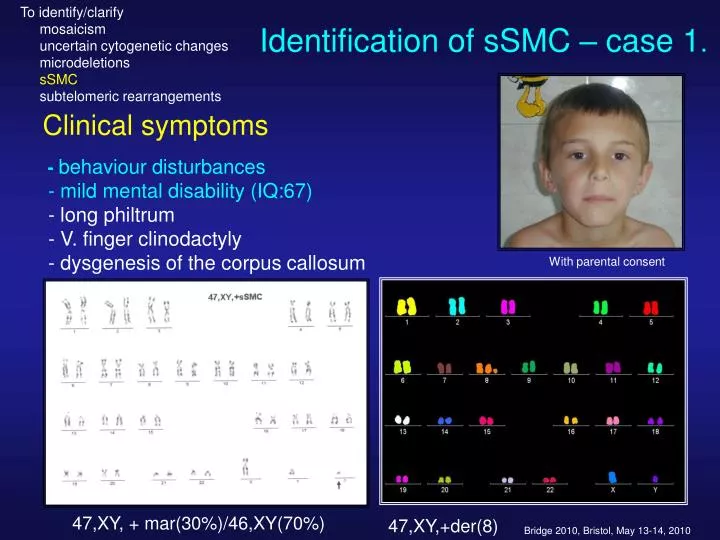
To identify/clarify mosaicism uncertain cytogenetic changes microdeletions sSMC subtelomeric rearrangements. Identification of sSMC – case 1 . Clinical symptoms - behaviour disturbances - mild mental disability (IQ:67) - long philtrum

E N D
To identify/clarify mosaicism uncertain cytogenetic changes microdeletions sSMC subtelomeric rearrangements Identification of sSMC – case 1. Clinical symptoms -behaviour disturbances - mild mental disability (IQ:67) - long philtrum - V. finger clinodactyly - dysgenesis of the corpus callosum With parental consent 47,XY, + mar(30%)/46,XY(70%) 47,XY,+der(8)
To identify/clarify mosaicism uncertain cytogenetic changes microdeletions sSMC subtelomeric rearrangements Detection of subtelomeric rearrangements. FISH: ToTelVysion Multi-color DNA probe Mixture • Subtelomeric regions are rich in genes • severe dysmorphic symptoms • 50% of cases proved to be inherited • (balanced carrier parent) • 5-10% of cases with idiopathic • severe mental disability (deletion, • duplication, translocation) • Some of them are associated with specific • phenotype (1p, 4p, 5p, 9q, 17p, 22q • Probes: Vysis ToTelVysion Probe Panel: • mixture of 15 individual „ready-to-use” direct • labelled probes specific for telomeric regions • of the p and q arms of all chromosomes • (excepted the p arms of acrocentric • chromosomes)
Detection of subtelomeric rearrangement – cases 1.-2. • Clinical symptoms • - Hypotonia • - Eye: Retinitis pigmentosa, decoloration • of papilla, nystagmus, div. strabism • - Polydactyly • Facial dysmorphy: depressed premaxillary • region, hypertelorism, ptosis, small, low set • ears, downward slanting mouth • - Severe mental disability • - Delayed psychomotoric development 8p subtelomeric deletion; 12p subtelomeric region trisomy in unbalanced translocation With parental consent.
Array Comparative genomic hybridisation (CGH) Advantages • to identify the exact breakpoints, localization of affected genes • to detect microdeletions, microduplications, polymorphisms of pathological importance • to study genotype – phenotype correlations
Complex glycerol kinase deficiency (CGKD): microdeletion of Xp21 Specific dystrophin gene array analysis Sample 3: 114/1 affected boy at contiguous gene deletion syndrome (Score dystrophin gene: -0,536; Score neighbouring genes: -0,742)
Complex glycerol kinase deficiency (CGKD): Xp21 microdeletion • dystrophin gene deletion by MLPA Neighbouring genes: • GK gene: Xp21.1 – glycerol kinase deficiency • NR0B1 gene: Xp21.3-21.2 – adrenal hypoplasia • Il1RAP gene: Xp22.1-21.3 – mental disability • RPGR gene: Accurate breakpoints Genotype-phenotype correlation
Genetic diagnostic approaches Known chromosomal syndromes, syndromes of unknown origin, mental disability, dysmorphic symptoms Chromosome analysis Numerical, structural aberrations Uncertain Normal karyotype Further steps delineated by the phenotype Chromosome aberration is compatible with the clinical features Fine chromosome alterations: markers’ identification, sSMC, microdeletion syndromes, subtelomeric rearrangements Syndromes of unknown origin 3. Syndrome identification: „search” >5600 Known monogenic syndromes: Fra-X, chondrodysplasia, syndromic craniosynostoses etc. FISH, MFISH, CGH Molecular genetic tetsts (mutation analysis: PCR, sequencing, RFLP etc.)
Molecular diagnosis of monogenic disorders • Diseases studied: • Fragile–X: FMR1 gene –Southern blot • Craniosynostoses: Apert, Crouzon, Pfeiffer • FGFR2 gene – PCR, sequencing, • RFLP • Achondroplasia: FGFR 3gene - RFLP • Hypochondroplasia: FGFR 2, 3 genes – • sequencing • Lissencephaly:LIS1 gene – FISH, sequencing
IgII IgI IgIII TM A TK1 L TK2 Apert syndrome: AD – FGFR2 mutation With parental consent Ser252Trp Pro253Arg: 99% Clinical symptoms Craniofacial dysmorphy: clover shaped cranium, high forehead,flat occiput, hypertelorism, deep nasal root, small, beaked nose, cleft palate Syndactyly, small muscular VSD
Genetic study DNA isolation Amplification of exon 8 in FGFR2 gene using PCR technique Sequencing, RFLP analysis (MboI) c.C755G ( Ser252Trp) mutation Healthy E B M 300 250 200 172 150 153 118 100 50 54 Patient
Apert syndrome 2. With parental consent FGFR2 c.C758G (p.Pro253Arg) Gain of function: Increased ligand-binding capacity Loss of ligand-binding specificity Genotype-phenotype correlation: Ser252Trp:cleft palate more common better recovery after operation Pro253Arg: more severe syndactyly
Further genetic tests to detect gene mutations performed in collaboration inside Hungary achondroplasia Apert sy DMP Rett sy FGFR2 FGFR3 dystrophin MECP2 PKU, CF, CAH, Werdnig-Hoffmann, Smith-Lemli-Opitz, glycosilation disturbances, haemophilia, Russel-Silver, Lysosomal storage diseases (Gaucher, Fabry, MPS I. Niemann-Pick, Pompe) , etc. • Significance: • to avoid unncessary • investigations • prognosis, • praenatal diagnosis • praesymptomatic diagnosis • prevent recurrence ORPHANET
Genetic diagnostic approaches Known chromosomal syndrome, syndromes of unknown origin, mental disability, dysmorphic symptoms Chromosome analysis Uncertain Normal karyotype Numerical, structural aberrations Further steps delineated by the phenotype Chromosome aberration is compatible with the clinical picture Fine chromosome alterations: markers’ identification, sSMC, microdeletion syndromes, subtelomeric rearrengements Syndromes of unknown origin 4. Syndrome identification: „search” >5600 Known monogenic syndromes: Fra-X, achondroplasia, Apert sy. etc. FISH, MFISH, CGH Molecular genetic tetsts (mutation analysis: PCR, sequencing etc.)
Diagnosis: Ohdo syndrome Diagnostic criteria: • blepharophimosis • ptosis • external acustic meatus stenosis +/- deafness • hypotonia • mental disability Photos are shown with parental consent
Magnetic resonance Imaging B=0 FA TRACE Standard MRI vs. Neuroimaging: T1 and T2 weighed MRI revealed no organic alteration in the brain, while fractional anisotropy and fiber trajectory imaging diclosed a demyelinated area close to the anterior horn of the ventricle causing a complete „communication” blockade
CONCLUSION • To identify the genetic background of congenital genetic disorders the whole spectrum of currently available genetic tests have to be applied according to a proper algorythm • The genetic diagnosis (etiology) determines the syndrome-specific additional symptoms as well as pattern and severity of intellectual disability. • The genetic diagnosis has to be considered when developing the early intervention, rehabilitation and preventive measurements.
Clinical Genetic Center, University of Debrecen Director of working group: Éva Oláh, MD, D.Sc. Members: Cytogenetics:Erzsébet Balogh, biol. PhD. FISH, PCR: Anikó Ujfalusi, MD. PhD Beáta Bessenyei, mol. biol. Syndrome diagnosis: Katalin Szakszon, MD. Gabriella P. Szabo, MD. Technical assistants: Mrs. Gábor Orvos Mrs. Ferenc Bodnár Ms. Éva Nagy PhD students: Attila Mohánszky, mol. biol. GabriellaP. Szabó, MD. Katalin Szakszon, MD. Ivett Körhegyi, MD. :Student Scientific Circle: students