Download
1 / 1
10 likes | 123 Views
Investigating genetic susceptibility of congenital polycythemia through shared genomic segment and linkage analysis in a unique extended family Karen Curtin, Nicola J Camp, Felipe R Lorenzo, Sabina Swierczek, Josef T Prchal

E N D
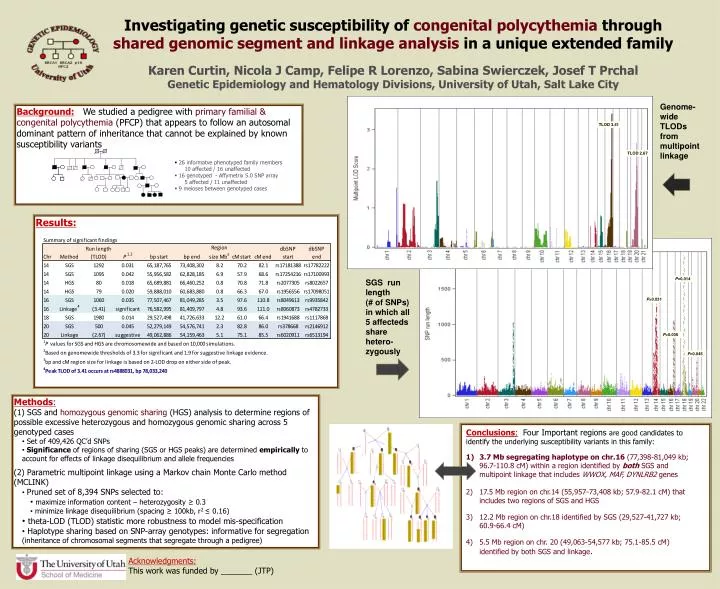
Investigating genetic susceptibility of congenital polycythemia through shared genomic segment and linkage analysis in a unique extended family Karen Curtin, Nicola J Camp, Felipe R Lorenzo, Sabina Swierczek, Josef T Prchal Genetic Epidemiology and Hematology Divisions, University of Utah, Salt Lake City Genome-wide TLODs from multipoint linkage Background: We studied a pedigree with primary familial & congenital polycythemia (PFCP) that appears to follow an autosomal dominant pattern of inheritance that cannot be explained by known susceptibility variants TLOD 3.41 TLOD 2.67 • 26 informatve phenotyped family members • 10 affected / 16 unaffected • 16 genotyped - Affymetrix 5.0 SNP array • 5 affected / 11 unaffected • 9 meioses between genotyped cases Results: P=0.014 SGS run length (# of SNPs) in which all 5 affecteds share hetero-zygously P=0.031 P=0.035 P=0.045 • Methods: • (1) SGS and homozygous genomic sharing (HGS) analysis to determine regions of possible excessive heterozygous and homozygous genomic sharing across 5 genotyped cases • Set of 409,426 QC’d SNPs • Significance of regions of sharing (SGS or HGS peaks) are determined empirically to account for effects of linkage disequilibrium and allele frequencies • (2) Parametric multipoint linkage using a Markov chain Monte Carlo method (MCLINK) • Pruned set of 8,394 SNPs selected to: • maximize information content – heterozygosity ≥ 0.3 • minimize linkage disequilibrium (spacing ≥ 100kb, r2 ≤ 0.16) • theta-LOD (TLOD) statistic more robustness to model mis-specification • Haplotype sharing based on SNP-array genotypes: informative for segregation (inheritance of chromosomal segments that segregate through a pedigree) • Conclusions: Four Important regions are good candidates to identify the underlying susceptibility variants in this family: • 3.7 Mb segregating haplotype on chr.16 (77,398-81,049 kb; 96.7-110.8 cM) within a region identified by both SGS and multipoint linkage that includes WWOX, MAF, DYNLRB2 genes • 17.5 Mb region on chr.14 (55,957-73,408 kb; 57.9-82.1 cM) that includes two regions of SGS and HGS • 12.2 Mb region on chr.18 identified by SGS (29,527-41,727 kb; 60.9-66.4 cM) • 5.5 Mb region on chr. 20 (49,063-54,577 kb; 75.1-85.5 cM) identified by both SGS and linkage. Acknowledgments: This work was funded by _______ (JTP)





















