Download

1 / 24
250 likes | 509 Views
INDIAN INITIATIVE FOR TOMATO GENOME SEQUENCING Tomato Finishing Workshop. T. R. Sharma National Research Centre on Plant Biotechnology Indian Agricultural Research Institute New Delhi -110012 trsharma@nrcpb.org. Tomato Genome Sequencing Project. Spain. USA. USA. Italy. France.

E N D
INDIAN INITIATIVE FOR TOMATO GENOME SEQUENCING Tomato Finishing Workshop T. R. Sharma National Research Centre on Plant Biotechnology Indian Agricultural Research Institute New Delhi -110012 trsharma@nrcpb.org
Tomato Genome Sequencing Project Spain USA USA Italy France Japan India N.land USA Korea China UK
Sequence Type Capillary Sequencers ABI-3700 MegaBace-1000/4000 Collection of DNA Seq. data
Softwares Developed for Performing HTGS Analysis • rename - renames any number of files from ABI or MegaBACE generated format to St. Louis naming convention, • fsplit - splits a file containing multiple sequences in fasta format • fmerge - converts multiple fasta files into a single fasta file • coverage - calculates the depth of coverage of an assembly by the most stringent method • extract_reads – extracts all the reads from a particular contig or contigs in an assembly, • comhits - compares two blast outputs stored as text for common hit • confasta - converts a file of nucleotide sequences containing numbers and/or blank spaces into a sequence fasta file for doing BLAST search • format2xls - converts sequence fasta files to a tab delimited format • format2fasta - converts a database stored file into fasta format for further analysis • prefinish96 - an excel macro program which arranges templates in alphabetical order along with their custom primers in a 96 well format • prefinish384 - a similar excel macro program for template arrangement in 384 well format
Genome Sequences Types Submitted to GenBank A B E F H G C D Phase I 4 1 3 2 A B C D E F G H Phase II E E 1 2 3 4 Single clone area Gap Gap Single strand area Custom primers Multiple clone coverage on both strands E E 1 Phase III
Finishing DNA Sequences Finishing: is the process of polishing raw sequences, transforming the fragmented rough draft into long, continuous final product without breaks or errors. GOALS……….. • Resolve sequence ambiguities and discrepancies, such that the error rate is less than one in 10,000 bases. • Provide “double-stranded” coverage for every base: • minimum of two different clones • two different directions • two different chemistries • Achieve contiguity. • Delineate vector/insert junctions.
Finishing DNA Sequences -How Scan assembly to pick linker clones for Tn Seq custom oligo dye terminator reverse dye terminator special chem (dGTP) reactions custom oligo for BAC DNA sequencing PCR amplification of problem areas Software used: Consed which is a graphical tool for viewing and editing sequence assembly data : chromat_dir, phd_dir, edit_dir
Methods to resolve Seq. Gaps 1.Transposon method Linker clones • Identify linker clones • Perform trnasposon insertions • Transform DH10B cells • Pickup atleast 24 white colonies • Prepare template • Seq. all the templates • Add new Seq. data (New England BioLabs)
Methods to resolve Seq. problems 2.Custom primer method Poor quality region Identify problem areas Custom primer Design primers Seq. at least 3 shot gun clones spanning to the region With same/different chemistry Add new seq. data - Editing
Methods to resolve Seq. problems 3.PCR method Primers Contig 1 Contig 2 PCR amplification M 1 2 3 4 5 6 7 8 1 kb - Cleaning of PCR products Seq. of PCR products New reads Joining 2 contigs by PCR
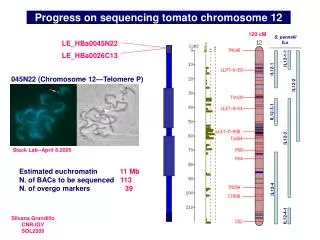
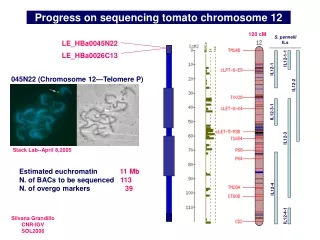
Sequencing Status, IITGS Phase 111 = 24 Phase 11 =25 Phase1 =10 Library =9 Total BACs Seq. = 68
BAC clones in Phase III (IITGS) Total Seq.=1.168MB
BAC clones in Phase III (IITGS) Total Seq.=1.283MB
BAC clones on other Chromosomes / Redundant BAC Clones Total Seq.=631kb Total Seq.=3.082MB
Aligned region showing single base mismatch in C05SLm0050C14 consensus
Approach to solve the misassembly in C05SLm0050C14 • Manually re-arranging reads on basis of: • Read-pair information of sub-clones. • PCR of different regions within the BAC to reconfirm assembly. • Digestion pattern of BAC obtained from six different restriction enzymes. • Sequence obtained after assembling individual sub-clones following transposition Current status of C05SLm0050C14 Region yet to be resolved
Misassembly C05HBa0089M06
ACKNOWLEDGEMENTS All Members of Indian Tomato Genome Sequencing Group and DBT for Financial Assistance