Download

1 / 23
300 likes | 837 Views
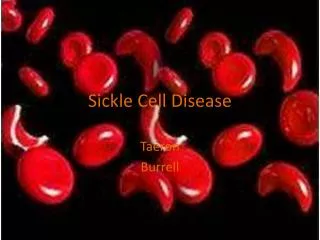
Sickle Cell Disease. Yohan Kim. Sickle Cell. First to described in 1904 by James B. Herrick. Found “elongated and sickle-shaped” red blood cells (RBCs) from a patient suffering from anemia

E N D
Sickle Cell Disease Yohan Kim
Sickle Cell • First to described in 1904 by James B. Herrick. • Found “elongated and sickle-shaped” red blood cells (RBCs) from a patient suffering from anemia • A multisystem disease, associated with episodes of acute illness and progressive organ damage • Most common in African heredity population • Occurs in about 1 of every 500 births • Clinical management of sickle cell disease is still basic and no drugs have been developed that specifically target the pathophysiology of this disease • Affects about 70,000 people in the US
Pathophysiology • 2 Types of Hemoglobin HbF(More O2), HbA(Less O2) • Newborn – 5:5~8:2, 6 months – 1:99 • Sickled hemoglobin is caused by a mutation of the gene of HbA • In the amino acid sequence valinetakes the place of glutamic acid at the sixth position. • HbA become C-shaped or sickled when the patient’s oxygen saturation is low.
Sickled RBC • Reduced flexibility impairs blood flow through vessels • Repeated de-oxygenation can damage the cells enough to make sickling permanent • short-lived (10 to 20 days instead of the normal 120 days)
Signs and symptoms • No clear symptoms shown • Indications may be visible during growth • Newborns usually of normal length and weight at birth • Develop a gradually increasing deficit in height and weight as they age • After time of the normal adolescent growth spurt, they begin to catch up with their normal peers. • (Girls 15 to 17, boys 18 to 19) • Delayed puberty also occurs.
Referral • Treatment of sickle cell crisis is usually symptomatic • Referring the patient immediately
Diagnostic test • Based on analysis of hemoglobin • Protein electrophoresis or chromatography • Hemoglobin mass spectrometry and DNA analysis increasingly used • Enable high-throughput testing.
Differential diagnosis • HbSS- inherited two sickle cell genes (S); one from each parent. • The most common type • HbSC - inherited a sickle cell gene (S) from one parent and a gene for abnormal hemoglobin (C) from the other • 20% to 30% of cases, usually a milder form of sickle cell disease. • HbS beta thalassemia - one sickle cell gene from one parent and one gene for beta thalassemia from the other parent. • Other: HbSD, HbSE, and HbSO-inherited one sickle cell gene and one gene for an abnormal type of hemoglobin • HbAS - sickle cell trait
Sickle cell trait • Not a disease, generally regarded as an asymptomatic condition. • When compared to controls, is no difference • Some physiological differences during exercise or at high altitudes • Following exercise -> increase in the number of sickled cells. • exertional sickling • The presence of sickled cells after physical exertion is even greater at higher altitudes
Sickle cell trait • Explanations for sudden deaths due to sickle cell anemia are not entirely clear. • Data indicate an association between sickle cell trait and sudden death there is no direct evidence of causation. • Dehydration, hyperthermia and acidosis associated with extreme physical exertion suggested • Results include rhabdomyolysis (muscle break-down), acute renal failure and coronary vasoconstriction
Treatment • Hydroxyurea (medication) • Oral efficacy and low toxic effects • Increase hemoglobin concentrations • Decreasing platelet and white cell counts • Changing expression of adhesion molecules • Nitric oxide generation
Treatment • Red blood cell transfusion • Corrects anemia • Decreases the percentage of sickled hemoglobin • Suppresses sickle synthesis, • Reduces polymerization. • Necessary if the hemoglobin concentration is high, or if rapid decrease in sickled hemoglobin percentage needed
Treatment • Hematopoietic stem cell transplantation • AKA bone marrow • The only cure for sickle cell disease • First bone marrow transplantation for sickle cell in 1983 on 8-year-old patient with leukemia and sickle cell disease • The bone marrow transplantation cured both diseases
Treatment • Hematopoietic stem cell transplantation • Indications • Stroke or CNS event lasting longer than 24 hours • Abnormal brain MRI • Elevated transcranial Doppler • Acute chest syndrome with recurrent hospitalizations • Two or more vaso-occlusive crises requiring hospitalizations • RBC alloimmunization
Treatment • Splenectomy • The spleen is the organ most often affected by sickle cell disease. • To prevent complication • Also reduces patients’ need for transfusion • Decreases splenic complications • Further eliminates pain
Complications • Acute pain - most common cause of hospital admission • Infection - major cause of morbidity and mortality • Stroke • Acute chest syndrome - second most common cause of hospital admission • Pulmonary hypertension • Heart disease • Renal complications - almost inevitable, hemoglobin polymerize in the renal medulla, because of the low oxygen
Prognosis • About 90% of patients survive to age 20 • Close to 50% survive beyond the fifth decade.
Prevention • No methods to prevention sickle cell disease • Screening and diagnosing sickle cell in advance will help prevent complications in the future • Engaging and educating patients before complications begin are also good ways of preventing complications.
Case study • A college football player with sickle cell trait (SCT) who collapsed minutes after running 16 successive sprints of 100 yards each at sea level. • The player, 19 year old, African American, was apparently healthy when running. • No exertional heat illness was present. • After collapsing, went into coma and developed fulminant rhabdomyolysis, profound lactic acidosis, acute myoglobinuric renal failure, refractory hyperkalemia, and disseminated intravascular coagulation. • Died about 15 hours after admission to hospital
Case study • Cause of death was acute exertionalrhabdomyolysis associated with SCT • appears that sickling can begin within 2-3 minutes of maximal exertion and can reach grave levels very soon • Being urged on by coaches can pose problem • Heat, dehydration, altitude, and asthma can increase the risk • Screening and simple precautions can prevent this unique syndrome and enable SCT athletes to thrive in their sports
References • 1. Redding-Lallinger R, Knoll C. Sickle cell disease--pathophysiology and treatment. Current Problems In Pediatric And Adolescent Health Care. 2006;36(10):346–376. • 2. Parrish M R, Morrison J C. Sickle cell crisis and pregnancy. Seminars in Perinatology. 2013;37(4):274–279. doi:10.1053/j.semperi.2013.04.006. • 3. Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. 2010;376(9757):2018–2031. doi:10.1016/S0140-6736(10)61029-X. • 4. Sheth S, Licursi M, Bhatia M. Sickle cell disease: time for a closer look at treatment options? British Journal Of Haematology. 2013;162(4):455–464. doi:10.1111/bjh.12413. • 5. Apanah S, Rizzolo D. Sickle cell disease: Taking a multidisciplinary approach. JAAPA: Journal of the American Academy of Physician Assistants (Lippincott Williams & Wilkins). 2013;26(8):28–33. doi:10.1097/01JAA.0000432497.24151.d4. • 6. Aloe A, Krishnamurti L, Kladny B. Testing of collegiate athletes for sickle cell trait: what we, as genetic counselors should know. Journal Of Genetic Counseling. 2011;20(4):337–340. doi:10.1007/s10897-011-9366-9. • 7. Anzalone ML, Green VS, Buja M, Sanchez LA, Harrykissoon RI, Eichner ER. Sickle Cell Trait and Fatal Rhabdomyolysis in Football Training: A Case Study. Medicine & Science in Sports & Exercise. 2010;42(1):3–7.