Download

1 / 37
400 likes | 922 Views
Biochemistry 412 Enzyme Kinetics I March 29 th , 2005. Reading: Mathews & van Holde, Biochemistry , Benjamin/Cummings Publishing Co., Redwood City, CA, pp. 341-364 in 1990 edition (or equivalent pages in a later edition) Other (optional) resources:

E N D
Biochemistry 412 Enzyme Kinetics I March 29th, 2005
Reading: Mathews & van Holde, Biochemistry, Benjamin/Cummings Publishing Co., Redwood City, CA, pp. 341-364 in 1990 edition (or equivalent pages in a later edition) Other (optional) resources: http://web.mit.edu/esgbio/www/eb/ebdir.html http://web.indstate.edu/thcme/mwking/enzyme-kinetics.html >>> And special thanks for this lecture goes to Dr. Gabriel Fenteany, Department of Chemistry, University of Illinois at Chicago (www.chem.uic.edu/fenteany/teaching/452), whose slides I liberally borrowed!
Enzymes are proteins that can accelerate biochemical reactions often by factors of 106 to 1012! This is much higher than chemical catalysts. Enzymes can be extremely specific in terms of reaction substrates and products. Enzymes catalyze reactions under mild conditions (e.g. pH 7.4, 37ºC). The catalytic activities of many enzymes can be regulated by allosteric effectors. Enzymes Are Uniquely Powerful Catalysts
For example: Triose Phosphate Isomerase
Irreversible First-Order Reactions k A B v = d[B]/dt = -d[A]/dt = k[A] (k = first-order rate constant (s-1)) Rearrange: d[A]/[A] = dln[A] = -kdt Integrate and express [A] as a function of time (t): [A]/[A]o = e –kt or [A]= [A]oe –kt ([A]o = initial concentration)
Reversible First-Order Reactions k1 A B v = -d[A]/dt = k1[A] - k-1[B] At equilibrium: k1[A]eq - k-1[B]eq = 0 [B]eq/[A]eq = k1/k-1 = Keq k-1
Second-Order Reactions k 2A P v = -d[A]/dt = k[A]2 A + B P v = -d[A]/dt = -d[B]/dt = k[A][B] (k = second-order rate constant (M-1s-1)) Change in [A] with time: 1/[A] = 1/[A]o + kt k Note: third-order reactions rare, fourth- and higher-order reactions unknown.
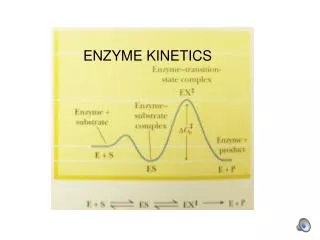
Free Energy Diagrams K = e –∆Gº/RT For A A‡ [A]‡/[A]o = e –∆Gº‡/RT [A]‡ = [A]o e –∆Gº‡/RT K = equilibrium constant ‡ = transition state [A]‡ = concentration of molecules having the activation energy [A]o = total concentration –∆Gº‡ = standard free energy change of activation (activation energy)
Relationship of Reaction Rate Constant to Activation Energy and Temperature: The Arrhenius Equation Reaction rate constant (k) determined by activation energy (∆Gº‡) and temperature (T) and proportional to frequency of forming product (Q = kBT/h, where kB = Boltzmann’s constant, h = Planck’s constant): k = Q e -G°‡/RT = (kBT/h) e -G°‡/RT (G = H - T S) k = Q e S°‡/R e -H°‡/RT k = Q´e -H°‡/RT (Q´ = Q e -S°‡/R) ln k = ln Q´ - H°‡/RT L-malate fumarate + H20 ln k
The Transition State Energy Barrier Opposes the Reaction in Both Directions K = k1/k-1 K = (Q e -G1°‡/RT)/(Q e -G-1°‡/RT) K = e -(G1°‡ -G-1°‡)/RT ∆Gº = G1º‡ - G-1º‡ K = e –∆Gº/RT Equilibrium constant K says nothing about rate of reaction, only free energy difference between final and initial states.
Effect of a Catalyst on Activation Energy • Catalysts do not affect GA (initial) or GB (final) and so do not affect overall free energy change (∆Gº = GB - GA) or equilibrium constant K. • Equilibrium concentrations of A and B still determined solely by overall free energy change. • Catalysts only affect ∆Gº‡, lowering the activation energy. • They accelerate both the forward and reverse reaction (increase kinetic rate constants k1 and k-1).
Q: How do enzymes work? A: A number of ways: “propinquity”, catalytic groups at active site, catalytic metals at active site, etc. (see assigned reading). >>> however, primitive enzymes may have behaved like catalytic antibodies, which can accelerate reactions merely by binding to and increasing the relative concentration of the transition state ([A]‡ = [A]o e –∆Gº‡/RT effect).
Oxidoreductases catalyze oxidation-reduction reactions. Transferases catalyze transfer of functional groups from one molecule to another. Hydrolases catalyze hydrolytic cleavage. Lyases catalyze removal of a group from or addition of a group to a double bond, or other cleavages involving electron rearrangement. Isomerases catalyze intramolecular rearrangement. Ligases catalyze reactions in which two molecules are joined. Types of Enzymes
Triose Phosphate Isomerase • Mutational analysis to study enzyme mechanism: • Site-directed mutagenesis - substitution mutation at specific position in sequence • Deletion mutation
Free Energy Barrier to the Glyceraldehyde-3-Phosphate <-> Dihydroxyacetone Phosphate Reaction Mutant in flexible loop that closes over active site.
The Effect of Substrate Concentration on Reaction Velocity Q: for a fixed amount of enzyme, what happens if you keep adding more and more substrate?
v = k2[ES], if this is the rate-limiting step* [Enzyme]total = [E]t = [E] + [ES] How to solve for [ES]? 1. Assume equilibrium, if k-1 >> k2: KS = k-1/k1 = [E][S]/[ES] or 2. Assume steady state: d[ES]/dt = 0 [*Note: v is always measured as an initial rate!] Michaelis-Menten Kinetics (1) E = free enzyme, S = substrate ES = enzyme-substrate complex P = product (Michaelis and Menten, 1913) (Briggs and Haldane, 1925)
Because of steady state assumption: d[ES]/dt = k-1[ES] + k2[ES] - k1[E][S] = 0 So: k1[E][S] = k-1[ES] + k2[ES] Rearranging: [ES] = (k1/(k-1 + k2))[E][S] Substituting (the “M” constant* = KM = (k-1 + k2)/k1): [ES] = ([E][S])/KM So:KM[ES] = [E][S] Michaelis-Menten Kinetics Continued (2) *Note: Briggs & Haldane came up with this, but they lost out when it came time to name things!
Michaelis-Menten Kinetics Continued (3) Substituting ([E] = [E]t - [ES]): KM[ES] = [E]t[S] - [ES][S] Rearranging:[ES](KM + [S]) = [E]t[S] So:[ES] = [E]t[S]/(KM + [S]) Now we can substitute for [ES] in the rate equation v = k2[ES], so…
The Michaelis-Menten Equation (4) v = k2[E]t[S]/(KM + [S]) or v = Vmax[S]/(KM + [S]) (since Vmax = k2[E]t)
At [S] « Km, Vo is proportional to [S]At [S] » Km, Vo = Vmax
E + S ES EP E + P v = kcat[E]t[S]/(KM + [S]) Multi-step Reactions k2 k3 k1 k-1 (kcat = general rate constant that incorporates k2 andk3)
kcat, KM, and kcat/Km: Catalytic Efficiency => “Perfect enzyme” Diffusion-controlled limit: 108-109 M-1s-1 Substrate preferences for chymotrypsin