Download
1 / 16
160 likes | 209 Views
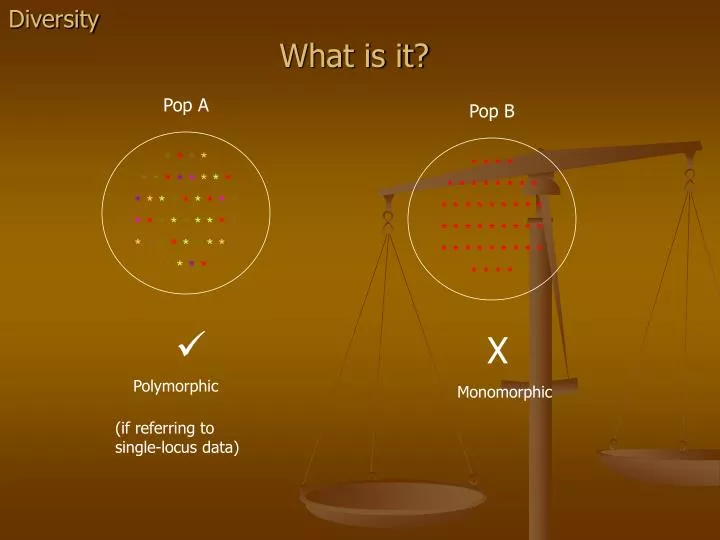
Diversity. What is it?. Pop A. Pop B. * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * *. * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * *. X. Polymorphic. Monomorphic. (if referring to single-locus data).

E N D
Diversity What is it? Pop A Pop B **** ******** ********* ****** *** ********* **** * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * X Polymorphic Monomorphic (if referring to single-locus data)
Diversity Why is it important? • Diversity is a reflection of • 1) Demographic history • 2) Mutational processes Almost without exception, when comparing two populations, we can assume that mutational processes are the same in both pops. Hence differences in diversity indicate differences in demographic history
Diversity TMRCA dictates diversity Time More diversity Large sum of branch lengths More time for mutations to accumulate Less diversity Small sum of branch lengths Less time for mutations to accumulate Large N (constant through time) No bottlenecks (N varies through time) Recent admixture Factors which increase TMRCA?…
Diversity How do we measure diversity? Problem: depends on sample size Despite appearing more complicated, has the advantage of interpretability Hence, finding differences in diversity can give us clues about differences in the demographic history of two populations So, how do we measure diversity? Depends on what we’re measuring. The simplest data are simple categories: Allele A, B, C, D etc. Even here, there is more than one way to measure diversity - Number of different alleles - Genetic diversity, h
Diversity Allele Frequency h interpretation I (Probability) A pA pA2 = probability that 2 randomly-chosen chromosomes are both AA B pB pB2 = probability that 2 randomly-chosen chromosomes are both BB C pC pC2 = probability that 2 randomly-chosen chromosomes are both CC D pD pD2 = probability that 2 randomly-chosen chromosomes are both DD h is the probability that two chromosomes picked at random from the population will be different (using the given genetic markers)
Diversity Allele Frequency h interpretation I (Probability) A 0.3 0.09 = probability that 2 randomly-chosen chromosomes are both AA B 0.2 0.04 = probability that 2 randomly-chosen chromosomes are both BB C 0.1 0.01 = probability that 2 randomly-chosen chromosomes are both CC D 0.4 0.16 = probability that 2 randomly-chosen chromosomes are both DD h is the probability that two chromosomes picked at random from the population will be different (using the given genetic markers) e.g…. In diploid systems, chromosomes naturally come in pairs. Here, h is also the “expected heterozygosity” – i.e. the expected frequency of heterozygotes if alleles joined at random (Hardy Weinberg Equilibrium)
Diversity h interpretation II (Variance) You may wonder why we use h in haploid systems, when chromosomes do not come naturally in pairs The answer is that h is still a good measure of diversity, and that thinking about pairs of chromosomes is still a natural way to think about the problem h is twice the “within-population variance”, when defined as follows…
Diversity Variance Deviation from the mean One example value of X from its distribution In statistics, the most widely used measure of diversity is variance (Note: standard deviation is derived from the variance with a 1-to-1 correspondence, so mathematically contains the same information (it is the square root of the variance)) X E[X] Variance is the expected squared deviation from the mean Var(X) = E[ (X – E[X])2 ] A little-known fact is the variance is also half the expected squared difference between 2 randomly-sampled X values Var(X) = E[ (X –X’)2 ]/2 = E[diff 2]/2
Diversity h interpretation II (Variance) Going back to diversity in a population, let us define diff=0 if 2 chromos are the same, and define diff=1 if 2 chromos are different What is E[ diff 2 ] for 2 randomly-drawn chromosomes? = Fr(same) x 02 + Fr(different) x 12 Hence, by defining variance in terms of difference between 2 objects, and defining diff=0 for ‘same’ and diff=1 for ‘different’, we gain a mathematical 1-to-1 relationship between h and variance h = Fr(different) = E[ diff 2 ] = 2*variance This is more nifty than it may at first appear, because variance is a concept normally applied to a scalar variable X, whereas h applies to a vector of frequency variables p1, p2, p3 … pm (where m is the number of different alleles in the population)
Diversity Estimating h By definition, Where p i is the true population frequency of Allele i An obvious estimate of h is therefore: Hat means this is an estimate But this estimate is biased – i.e In practice, we never know p i , only an estimate x i based on sample counts: x i = a i /n where a i = number of Allele i in sample and n = total sample size
Diversity Deriving an unbiased estimate of h The following is is not a full explanation, but hopefully will give the gist of it Remember that h can be derived by thinking about picking 2 chromosomes at random from the true population The true population, for this purpose, is assumed to be infinite so that it is impossible to pick the same chromosome twice To mimic this situation in the sample we have taken, we must arrange things so that the two chromosomes are picked without replacement from the sample
Diversity Deriving an unbiased estimate of h Some algebra results in: Adjust to avoid self-matches… Each number in the grid below represents a different chromosome in the sample a1 = 3 a2 = 3 a3 = 4 Area of “box” = n 2 Unadjusted frequency of ‘same’ matches: (a12 + a22 + a32)/n 2 Adjusted frequency of ‘same’ matches: (a12 + a22 + a32 – n) / (n 2 – n) Adjusted frequency of ‘different’ matches: 1 – (a12 + a22 + a32 – n) / (n 2 – n) 1 2 34 5 67 8 9 n a1 a2 1 2 34 5 67 8 9 n a3
Diversity The sampling distribution of hunb ^ ‘True’ h has no variance – there is only one unique value for each population Estimatedh does have a variance – you will get a slightly different value every time you sample n chromosomes from the population, because the sample will be different ‘true’ h = 0.9
Diversity The sampling distribution of hbiased ^ ‘true’ h = 0.9
Diversity For each resample, we calculate and use the values over many resamples to build up the bootstrap distribution for ^ Estimating the sampling distribution of h by bootstrapping What is bootstrapping? In bootstrapping, we assume that the estimated allele frequencies x iARE the ‘true‘ frequencies p i We now resample “fake” samples of size n from this imaginary population, lots of times
Diversity The bootstrap distribution of hunb ^ Because bootstrapping resamples the sample, and not the population, the resulting bootstrap distribution is biased In fact, there is no absolutely watertight way of testing for the difference between two h values. For this reason, I use a double-conservative procedure (see http://www.tcga.ucl.ac.uk/software) ‘true’ h = 0.9
