Download
1 / 13
130 likes | 268 Views
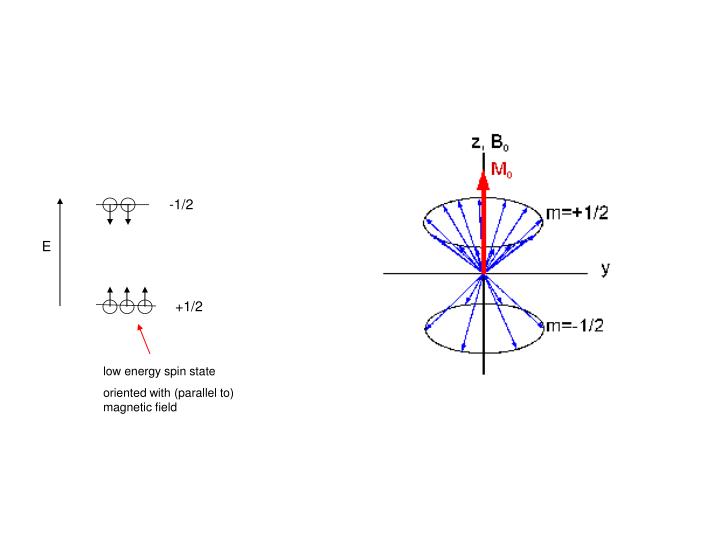
-1/2. E. +1/2. low energy spin state oriented with (parallel to) magnetic field. Two Dimensional NMR. 90 o x. 1D “pulse and acquire”. FT (t1). t1. relaxation delay. 90 o x 90 o x. t2. t1 delay. FT (t1,t2).

E N D
-1/2 E +1/2 low energy spin state oriented with (parallel to) magnetic field
Two Dimensional NMR 90o x 1D “pulse and acquire” FT (t1) t1 relaxation delay 90o x 90o x t2 t1 delay FT (t1,t2) In a 2D experiment FIDs are acquired for (perhaps 512) incremented values of the t1 delay (to generate a second frequency dimension). The data are Fourier transformed with respect to both t1 and t2. General rule: all 2D NMR spectra contain the 1D spectrum along the diagonal. Off-diagonal signals indicate a correlation (coupling or NOE depending on the type of spectrum) between 2 protons.
For example: The COSY spectrum below illustrates sequential spin coupling connectivities between protons within an amino acid. In the TOCSY spectrum each NH group is “labelled” with the frequencies of all the other protons in its amino acid side chain. COSY and TOCSY are useful for spectral assignment. In a NOESY spectrum, off diagonal signals correlate positions of protons that are within 5 angstroms. Very useful for structure determination. Note that COSY is the 2-dimensional analogue of spin decoupling NOESY is the 2-dimensional analogue of the difference NOE experiment COSY TOCSY
NOESY off diagonal “crosspeaks” correlate the chemical shifts of pairs of protons that are close in space (< 5 angstroms). semi-quantitative constraints on separation: weak 1.5 - 5 Å medium 1.5 - 3.5 Å strong 1.5 - 2.5 Å 0 ppm 2 4 6 8 red lines highlight a weak NOE between protons at 8.3 and 3.7 ppm. Wherever these protons are in the protein sequence, they must be close in space (< 5 Å) in the folded protein. 10 10 8 6 4 2 ppm backbone NHaromaticCHaCH b,g,d,e methyl
ppm 2 4 6 8 10 10 8 6 4 2 ppm The fingerprint region of a 2D 1H,1H spectrum contains coupling (COSY) or spatial (NOESY) correlations between backbone NH signals and backbone CHa signals. The fingerprint region of COSY and NOESY are used together for sequental assignment of the NMR spectrum. CHa NH In a COSY spectrum the fingerprint region contains one cross peak for each amino acid (I.e. intramolecular NH-CHa signal), except for proline (no NH) and glycine (2 CHa; i.e. 2 cross peaks for gly). NH signals here CHa signals
sequential assignment COSY CHa a CHa b through space correlation (NOESY) through bond correlation (COSY) NOESY CHa a CHa b for any backbone torsion angles found in a protein (f, ), either the distance dNiNi-1 or the distance dNiai-1 is short enough (<3.5 Å) to give an NOE effect. NHa NHb
must by Gly (only amino acid with 2 CHa) COSY NOESY Gly CHa’s here Ala CHa here not in COSY-probably Gly NH-Ala CHa NOE Gly NH here also in COSY (therefore intraresidue NHCHa NOE): i.e. Ala CHa - Ala NH Ala1-Gly2-Ser3-Leu4-Gln5-Asp6- L4 S3 Q5 G2 D6 A1 CHa G2 L4 A1 Q5 S3 D6 NH
Structure calculation Use computer energy calculation (commonly simulated annealing using Xplor) to determine structures that satisfy the NMR constraints NMR-derived structural constraints NOE’s (by far the most important) these are semi-quantitated e.g. weak NOE - constraint 1.5 - 5 Å medium - constraint 1.5 - 3.5 Å strong - constraint 1.5 - 2.5 Å backbone dihedral angles (phi) from NHCHa coupling constant hydrogen bonds provide distance constraints on N-H--O=C) from hydrogen exchange measurements (novel constraints: orientational information from residual dipolar couplings in partially aligned samples) Nature of NMR structure Since no single structure is defined by the full constraint set, a family of structures is calculated. These are usually presented as overlays of backbone traces to give a visual impression of the precision of the structure. Would generally like 10 NOE constraints per residue. NMR structures are usually well-defined in the backbone (rmsd around 0.2-0.4 angstroms) and less well defined in the side chains (0.5-0.8 angstrom rmsd). Investigation of ill-defined regions can be made using 15N relaxation measurements to distinguish between “floppy” structure and those lacking sufficient constraints. The more constraints, the better (more precise and hopefully more accurate) the structure.
CH CH 2 2 O O N C C N C C N H H H H H Three (and more)-dimensional NMR 15N- and 15N,13C-labelling allows structure determination of larger proteins (by simplifying increasing complex spectra, by alleviating inherent limitations due to slower “tumbling” of proteins in solution, and by providing improved assignment strategies). Structure determination still relies on identification of NOE’s. 15N NOESY HSQC: 3D spectra in which H-H NOESY information is resolved into separate planes in the 3d dimension according to the frequency of the amide 15N. Likewise for 15N TOCSY HSQC. CH CH 2 2 O O NCCNCCN H H H H H HNCA: 3D triple resonance spectrum in which cross peaks in 3D correlate the frequency of 15N, its directly attached 1H and the 13Ca of both its own and preceding amino acid. Backbone can be assigned directly by “walking” along the peptide backbone. And there are many other examples (e.g. see Dr. Schirra’s web site)
NMR in the Post Genomic Era (Structural Proteomics) 30,000 genes in human genome (35 - 40% membrane proteins) perhaps 3,000-10,000 different protein folds (identification of all folds plus homology modelling should allow fitting a structure to all gene products even without structure determination requires: increasing efficiency/throughput of structure determination (even low resolution determination of protein fold is valuable) improved structural determination of high MW proteins and membrane proteins actual/potential sources of improvement: 15N/13C labelling expression of isolated domains (most protein domains 50-150 amino acids) see Campbell & Downing paper on background reading list) total or partial deuteration of proteins (reduce line broadening from 1H-1H dipolar interactions) dipolar couplings: (partial orientation of protein in solution: determine 15N-1H and 13C-1H bond orientations) accurate alignment of 2iary structure elements in poorly constrained structures TROSY: novel pulse sequences that dramatically reduce line broadening at very high fields (see Riek et al. paper on reading list)
M. thermoautotrophicum Escherichia coli Sacchromyces cerevisiae Thermotoga maritima myxoma virus proteins from 513 ORF having predicted MW < 23kD expressed as His-tagged fusions in E. coli in 15N media. goodpromising mostly unfolded poor loss of protein upon concentration
