Download

1 / 23
470 likes | 2.11k Views
Neurodegenerative disease models. Neurodegeneration models in C.elegans. The development of transgenic models, has been intensively used for investigating neurodegenerative disorders such as Alzheimer's disease, Parkinson's disease, Huntington's disease and tauopathies.

E N D
Neurodegeneration models in C.elegans • The development of transgenic models, has been intensively used for investigating neurodegenerative disorders such as Alzheimer's disease, Parkinson's disease, Huntington's disease and tauopathies. • In some of these cases, the underlying genes do not have readily recognizable orthologues in C. elegans. • Nevertheless, conserved responses or interactions can often be detected.
Neurodegeneration models in C.elegans Kaletta et al. Nature Reviews Drug Discovery advance online publication; published online 21 April 2006 | doi:10.1038/nrd2031
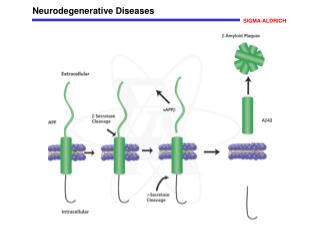
Alzheimer’s disease model • The Aβ1-42 fragment is the main neurotoxic peptide generated from APP • Invertebrate APP-like genes do not include the region encoding the neurotoxic Aβ1-42 fragment and, therefore, direct disease modelling based on endogenous APP cleavage is not possible. • The most developed invertebrate model of Aβ1-42 toxicity involves intracellular expression of a human Aβ1-42 fragment in the C. elegans bodywall muscle, leading to adult-onset progressive paralysis and shortened lifespan • In the nematode model, Aβ1-42 forms cytoplasmic β -amyloid with some properties similar to those found in diseased human brains
Model of age-related protection against proteotoxicity • DAF-2 represses two downstream pathways: transcription factor HSF-1 and transcription factor DAF-16. • Both provide protection against proteotoxicity of the amyloid 42 peptide, an aggregation-prone peptide that can spontaneously form small toxic aggregates. • The default pathway, regulated by HSF-1, identifies and breaks apart toxic aggregates. • When the HSF-1 machinery is overloaded, however, a molecular apparatus regulated by DAF-16 leads to formation of less toxic high-molecular-weight aggregates. Cohen et al. 2006
Parkinson’s disease • PD is the most common neurodegenerative movement disorder. • Approximately 1% of the population older than 65 years suffers from this slowly progressive neurodegenerative disease; 95% of PD cases are sporadic. • The symptoms of PD are caused by selective and progressive degeneration of pigmented dopaminergic (DA) neurons in the substantia nigra pars compacta. • Current treatments, such as administration of L-DOPA to produce dopamine, are only symptomatic and do not stop or delay the progressive loss of neurons. • In fact, some studies have suggested that oxidative injury via dopamine may lead to further neuronal damage. • 7 known genetic loci. • PARK1 is mutation in the gene for α-synuclein (synaptic vesicle fusion). • PARK2 is mutation in parkin, the ubiquitin ligase for a-synuclein. • The α-synuclein protein is also seen in Alzheimer’s plaques.
C. elegans Parkinson’s disease models • C. elegans has 302 neurons of which 8 are dopaminergic • Expression of a human gene encoding the PD-associated protein, alpha-synuclein, in C. elegans neurons results in dosage and age-dependent neurodegeneration • -synuclein-induced dopaminergic neurodegeneration could be rescued in these animals by torsinA, a protein with molecular chaperone activity http://www.jove.com/index/details.stp?ID=835
Phenotypes in a worm model of human PD postural and movement deficits loss of neurons Lakso et al., 2003
C. elegans Parkinson’s disease models • Impact of 6-hydroxydopamine (6-OHDA) on dopamine (DA) neurons in C. elegans • intact green-fluorescing DA neurons in an untreated worm • similar worm three days after exposure to 6-OHDA • The partial loss of green fluorescence is indicative of progressive death of the dopamine neurons - due to destructive reactive oxygen species produced by 6-OHDA Nass et al., 2002 PNAS
6-OHDA has been found in brain and urine samples of PD patients • DA transporter (DAT-1) in C. elegans is essentially the same, structurally and genetically, as the one found in mammals • Blocking DAT-1 function through drugs or by genetic disruption eliminates 6-OHDA-sensitivity in the worms • These results suggest that the toxic effect of 6-OHDA occurs after it enters the DA neuron. 6-hydroxydopamine
Huntington disease (HD) • HD affects medium sized spiny neurons • Uncontrollable growth of dendrites causes neurons to malfunction • Parts of the brain affects include the cortex and basal ganglia, particular the caudate nucleus and the striatum • Symptoms: Involuntary chorei-form (dance-like) movements, psychological change, dementia • Treatment aimed at symptoms, drugs that are helpful at one stage may not work at another stage • Death usually occurs 15 to 20 years after onset of symptoms • People die of complications to HD not HD itself - choking, infections, heart failure, pneumonia
HD is a monogenic disease • The disease is associated with increases in the length of a CAG triplet repeat present in a gene called 'huntingtin' (IT15) located on chromosome 4p16. • Wild-type huntingtin has a role in membrane trafficking in the cytoplasm and is also involved in microtubule-based axonal transport. • The range of CAG repeat numbers is 9 to 37 in normal individuals and 37 to 86 in HD patients. Huntingtin (Htt) domain model indicating the location of the polyQ repeat, proline-rich domain (PRD) and HEAT repeats. Selected interaction partners are listed for the polyQ and PRD region as well as the N-terminal region, including the first HEAT repeat section
Mutant huntingtin binds to synaptic vesicles with higher affinity than does wild-type huntingtin and inhibits the uptake of glutamate by synaptic vesicles, suggesting that it might be able to affect synaptic homeostasis directly. • Aggregates containing mutant huntingtin are usually located in the nucleus and cytoplasm, but they can also appear in the axon and nerve terminals.
Immunoblot showing anti-GFP immunoreaction in C. elegans protein extracts using 3–4-day-old animals expressing different lengths of GFP–Htt–polyQ proteins • GFP fluorescence micrographs of young adult (3–4 days old) C. elegans expressing different lengths of GFP–polyQ fusion proteins. Note that GFP fluorescence is mainly localized to the body wall muscle cells. Also, note that more compact foci form with increasing number of polyQ repeats expressed Wang, H. et al. 2006 Hum. Mol. Genet.
C) Higher magnification showing the body wall of young adult C. elegans expressing different lengths of GFP–polyQ fusion proteins D) Motility assay measured as body bends per minute in wild-type and various transgenic lines of adult C. elegans. Note that the rate of movement decreases with increasing length of polyQ repeats.
Ion-channel-mediated neurotoxicity • If neurons are deprived of oxygenand the ATP stores drop, excess excitatory neurotransmitter glutamate is released into the synapse. • Glutamate transporters, which usually rapidly clear synaptic glutamate, also fail to function appropriately in these conditions, leading to hyperactivation of glutamate-gated ion channels, excess ion influx, neuronal swelling and death. • In C. elegans, a mutant acetylcholine receptor Ca2+ channel DEG-3(d), and hyperactivated mutant variants of the degenerinNa+ channels cause neuronal swelling and death • Transgenic expression of constitutively active GTPase-defective heterotrimeric G protein Gs can also induce necrotic-like neuronal death • Because G proteins are known to influence ion-channel activity, Gs might induce neuronal degeneration by ion-channel hyperactivation
Hyperactivated ion channels, such as the MEC-4(d) Na+ channel, the DEG-3(d) Ca2+ channel and possibly other channels that are affected if Gs is constitutively activated, conduct excess ions into neurons. • This might induce the secondary release of calcium through ryanodine receptor (UNC-68) or InsP3R receptor (ITR-1) from ER stores, where it is bound by the ER chaperone proteins calreticulin and calnexin • If intracellular calcium levels rise, specific calpain and aspartyl proteases are activated and the cell ultimately undergoes a necrotic death. • In mammalian models of ischaemia and excitotoxic cell death, as well as in human neurodegenerative disease, activation of Ca2+-activated calpain proteases is crucial for necrotic demise