Download

1 / 4
40 likes | 222 Views
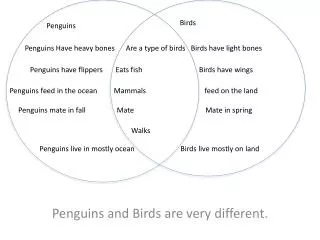
1. Genome Sequence (actg). 2. Proteome Sequence (amino acid). 3. HMM-based classification into structural fold families. Fe heme bound oxidative defense.

E N D
1. Genome Sequence (actg) 2. Proteome Sequence (amino acid) 3. HMM-based classification into structural fold families Fe heme bound oxidative defense 4. A “metallome” for each proteome is constructed using a manually curated annotation of the SCOP database. Includes structural and functional information Fe His bound vitamin metabolism Zn His bound carbon assimilation Introduction Chemical speciation modeling shows that Fe, Zn, Mn, and Co concentrations in an Archaean anoxic ocean, a Proterozoic euxinic ocean, and a Modern oxic ocean would have been quite different (Fig. 1). R.J.P. Williams and J.J.R. Frausto da Silva have long contended that these changes have had an indelible effect upon the evolution of life, particularly in the selection of elements for biological usage. Their theories further posit that this selective force will have left imprints in the genomes of organisms, though this has not been tested. Here we present the metal-binding structural contents of modern proteomes, as they are inferred from bioinformatics analysis of fully sequenced genomes. These results are reconciled with the theorized changes in global trace metal geochemistry. • Modern proteomes and putative “metallomic” imprints of ancient changes in geochemistry • Christopher L. Dupont1, Song Yang2, Brian Palenik1, Philip E. Bourne3 • Scripps Institution of Oceanography, University of California, San Diego • Department of Chemistry and Biochemistry, University of California, San Diego • San Diego Supercomputer Center and the Department of Pharmacology, University of California, San Diego • Contact: cdupont@ucsd.edu Fe binding folds: Oxygen and redox shifts Table 1: The seven most abundant Fe binding folds in each Superkingdom, along with the mode of Fe binding. Also shown is if O2 is present in reactions catalyzed by that fold. Essentially, Eukaryotic Fe binding folds are more likely to bind Fe by hemes or amino acids and also show an increased usage of oxygen. This is consistent with the hypothesis that Eukarya evolved in an oxic environment. The abundance of metal binding structures in a proteome adheres to a power law Figure 1: Theoretical levels of trace metals and oxygen in the deep ocean through Earth’s history. Whether the deep ocean became oxic or euxinic following the rise in atmospheric oxygen (~2.3 Gya) is debated, therefore both are shown (oxic ocean-solid lines, euxinic ocean-dashed lines). The trace metal concentrations are replotted from Saito et al, 2003. The phylogenetic tree symbols at the top of the figure show the theoretical periods of diversification for each Superkingdom. Figure 3: Panel A: Power law scaling for the abundance of metal binding domains. Each point is a discrete proteome of an Archaea (■), Bacteria (+), or Eukarya (o), with the number of Zn binding proteins on the Y-axis plotted against the total number of structural domains in a proteome, which is linear with genome size. Panel B: The slopes of the fitted power laws for Zn, Fe, Mn, and Co for each Superkingdom, which are evolutionary constants of proteome evolution (see below). The importance of “small class” Zn folds to Eukarya Figure 5: A: Log-log plot of the abundance of “small class” Zn binding folds in the proteomes for each Superkingdom. B: Venn diagram showing the distribution of the 53 unique small class Zn folds in each Superkingdom. The bottom set of numbers describe the distribution of small class Zn folds that occur in at least 50% of the proteomes in a given Superkingdom. Small class Zn folds are exemplified by Zn fingers and RING domains. They are believed to have originally evolved in Archaea. It seems unlikely that the observed diversification of Zn structures could occur in an environment low in Zn (Fig. 1). Methodology: Making the metallome Figure 2: Pathway of metallome construction. The results of steps 1, 2, and 3 are contained in the Superfamily database Step 4 is done using a manual annotation from the SCOP database. • Power Laws: fundamental constants in the evolution of proteomes • The power law is described by the function y = mxb. A slope of 1 indicates that a group of structural domains is in equilibrium with genome growth, while a slope > 1 indicates that the group of domains is being preferentially duplicated (or retained in the case of genome reductions). • The number of metal binding structural domains (nm)in a proteome of size pat any given time (t) are described by the generic equation: • nm (t) = (nm (0) / p(0)<am>/<a> )p(t)<am>/<a> • where <am> and <a> are time averages of the growth of a category and the entire proteome, respectively. • What does this mean? • The first term (blue) is defined by a common ancestor (time zero), and thus is the same for all proteomes in a given Superkingdom • The second term is the slope of our observed power laws, indicating that the abundances of Zn, Fe, Mn, and Co binding domains conform to Superkingdom-specific evolutionary constants, regardless of the evolutionary history of the organism. • Therefore: • The proteomes of the Prokarya have preferentially retained or recruited Fe and Co binding domains during increases or decreases in proteome size, respectively, while excluding Zn binding domains • Visa versa in the proteomes of Eukarya • Potential methodological biases • Unknown folds: The results from the Protein Structure Initiative suggest that there will be few novel metalloproteins of widespread distribution and high abundance • Genome Bias: Principal component analysis shows oxygen tolerance and environment have little effect upon the trends observed in Fig. 4. Phylogeny groupings are apparent however. Metallomes are very diverse Ubiquitous metal binding folds? Very few folds are found in all or most (>90%) proteomes. These include the tRNA synthases (Zn), Enolases (Mn), HemN (O2 independent coproporphyrin oxigenase), and HighPotentialIronProteins (HIPIP) Why are are the power laws different for each Superkingdom? Power laws are likely influenced by selective pressure. Qualitatively, the differences in the power law slopes describing Eukarya and Prokarya are similar to the shifts in trace metal geochemistry that occur with the rise in oceanic oxygen We hypothesize that they are the result of the environment of the last common ancestor in each Superkingdom This suggests that Eukarya evolved in an oxic environment, whereas the Prokarya evolved in anoxic enironmonts Do the metallomes contain further support this hypothesis? Figure 3: A quantile plot showing the percent of Bacterial proteomes each Fe-binding fold family occurs in (x). This plot also shows the average copy number of that fold family in the proteomes where it occurs (♦). Essentially, few Fe-binding folds are in most proteomes. Further, the widespread Fe-binding folds are not necessarily abundant. Similar trends are observed for Zn, Mn, and Co in all three Superkingdoms. • Conclusions • Metallomes have diverse compositions, yet the total abundances conform to evolutionary constants • These constants exhibit Superkingdom-specific differences consistent with ancient changes in geochemistry, a hypothesis further supported by the roles of Zn and Fe • These results provide genomic-based evidence for the theory of Anbar and Knoll that Eukaryotic diversification and oxygen-related changes in trace metal chemistry are linked Feb 1, 2008 Professional Development Series • References and Acknowledgements • Any work by JJR Frausto Da Silva and RJP Williams, Saito et al. 2003 Inorganica Chimica Acta 356: 308-318. Anbar and Knoll 2002 Science 297: 1137-1142, Van Nimwegen in Koonin et al. Power Laws. C.L.D. would like to thank the Princeton Center for Environmental Bioinorganic Chemistry and the ASEE (NDSEG fellowship) for funding; PEB is funded by NIH.
Our laboratory is very interested in scientific dissemination in the Web 2.0 era. To this end we have two major projects. • BioLit is the work of Dr. Lynn Fink and involves the integration of biological database content with the biological literature. We are using the complete corpus of the Public Library of Science journals (PLoS; www.plos.org) and the Protein Data Bank (PDB; www.pdb.org) as our prototype system. So for example, if you access a PLoS paper online describing a structure-function relationship, you can click on a figure in the paper and by accessing the associated structural data in the PDB bring up a view of the molecule that maps directly to that presented in the paper, rotate it, annotate it, and use it to further query the PDB and the associated literature. • SciVee is led by Apryl Bailey and involves Lynn Fink, John Matherly, Alex Ramos, Willy Suwanto and Ben Wilson. We refer to it as a YouTube for scientists. Check it out at http://scivee.tv • Kristine Briedis • Iowa State University, B.S. Genetics • Bioinformatics Graduate Program • 6th year PhD student • Using Structure Similarity to Search for New Human Protein Kinases Scientific Dissemination and Communication The proteasome is a multi subunit structure that degrades proteins. Protein degradation is an essential component of regulation because proteins can become misfolded, damaged, or unnecessary. Proteasomes and their homologs vary greatly in complexity. We searched 238 complete bacterial genomes for structures related to the proteasome, and found evidence of two novel groups of bacterial proteasomes. The first, which we name Anbu, is sparsely distributed among cyanobacteria and proteobacteria. We hypothesize that Anbu is an ancient proteasome. We also present evidence for a fourth type of bacterial proteasome found in a few β-proteobacteria, which we name β-proteobacteria proteasome homolog (BPH). Sequence and structural analysis show that Anbu and BPH are both distinct from known bacterial proteasomes, but have homologous structures. Anbu is encoded by one gene, so we postulate a duplication of Anbu created the 20s proteasome. We have found different combinations of Anbu, BPH, and HsIV within these bacterial genomes which raises questions about specialized protein degradation systems. • This project utilizes the EOL pipeline to identify new human kinases with its automated annotation tool, iGAP. In addition to traditional sequence alignment, the more conserved structural elements are considered when searching for remote homologs. This is achieved by comparing proteins to a comprehensive fold library to predict function and structure. • A novel protein kinase function for an Acyl-CoA dehydrogenase protein has been discovered with this process. This is potentially significant because kinases have been implicated in many diseases, including some forms of cancer, thus providing a new pharmaceutical target for therapy. We are interested in collaborations to further explore the role of this putative kinase. Email kbriedis@ucsd.edu for more information. The Bourne Laboratory http://www.sdsc.edu/pb This work is supported by NIH GM63208. Analysis of the Human Kinome Using Methods Including Fold Recognition Reveals Two Novel Kinases K.M. Briedis and P.E. Bourne PLoS ONE, Submitted. Our laboratory works in the general area of bioinformatics, with an emphasis on structural bioinformatics – the use of the complete corpus of macromolecular structure – proteins, DNA, RNA and complexes thereof to further our understanding of living systems. We believe that when studying living systems the devil is in the details, and in many cases structure affords those details. Our raw data are the Protein Data Bank (PDB) which we maintain for the worldwide community and is used by 10,000 scientists every day. Using these data we develop algorithms and methods in an attempt to improve our understanding of biology through computation. Here you will find the work of some of our students who study, for example, species differentiation based on protein fold content, prediction of sites of protein-protein interaction, prediction of binding sites across the druggable proteome, and the discovery of novel protein kinases within the human genome. We are committed to the free distribution of software and to open access to all our findings. • Song Yang • Beijing University, B.S. Chemistry • Department of Chemistry and Biochemistry • Graduated with PhD • Genome-wide Study of the Evolution of • Protein Domains I am interested in the evolutionary aspect of protein structures. Protein domain, the basic three-dimensional structural element of proteins, is stabilized by its intrinsic physical and chemical properties. Each domain has its own specific functions and occupies a particular sequence space thus resulting in its own evolutionary history. The study of the evolution of protein domains is not only an interesting topic, but further enhances our understanding of the sequence-structure-function relationship of proteins. Utilizing protein domains to address evolutionary problems and to study the evolution of protein domains themselves are two facets of the topic I am working on. The right hand side figure is a phylogenetic tree of 174 species across all three major kingdoms generated using protein domain content. My project identifies where and how proteins interact with each other using protein sequences and structures. We focus on exploiting the information extracted from 3D structures, which are expected to be very useful with the growing number of structures determined by structural genomics efforts. Structurally conserved residues, derived from multiple structure alignments, are combined with sequence profile and accessible surface area to predict protein-protein binding sites. The incorporation of structure conservation significantly improves the prediction performance. We are currently developing a prediction method to detect if two binding sites are interacting with each other. The ultimate goal of this project is to identify the binding sites of a protein and the corresponding binding site on the interacting protein partner. Exploiting Sequence and Structure Homologs to Identify Protein-Protein Binding Sites. J.L. Chung, W. Wang, and P.E. Bourne 2006 Proteins: Structure, Function and Bioinformatics 62(3) 630-640. This work is supported by the NIH grant 1P01GM63208-01A1 and 2T32 GM08326. • Jo-Lan Chung • National Taiwan University, B.S. - Chemistry 1999 • Department of Chemistry and Biochemistry • Graduated with PhD Exploiting Sequence and Structure Homologs to Identify Protein-protein Binding Sites Phylogeny Determined by Protein Domain Content. S. Yang, R.F. Doolittle, and P.E. Bourne. 2005 PNAS 102: 373-378 • Lei Xie, PhD • Researcher • Repositioning Existing Pharmaceuticals The PDB contains a significant number of major pharmaceuticals bound to their receptors. Lei Xie with Sarah Kinnings and Jian Wang, have developed a methodology for finding equivalent binding sites across what we define as the druggable proteome. At this time, we estimate this covers about 40% of all druggable targets. An equivalent binding site for a major pharmaceutical holds promise for either (a) explaining the side effects of existing drugs, or (b) using an existing drug (already approved) to treat a different condition. Thus far we have one example of each. Selective Estrogen Receptor Modulators (SERMs) are a class of drugs that include tamoxifen which are used in the treatment of breast cancer. This drug has significant side effects attributed to disruption in calcium homeostasis. We believe we have found the target of this epidemilogy, namely a Sacroplasmic Reticulum Ca2+ ion channel ATPase protein (SERCA). The challenge now is to design a modified SERM that has equal or better binding to estrogen receptors but less binding to SERCA. In a second experiment, we have established a Parkinson’s Disease drug which we believe will be very effective in the treatment of drug resistant tuberculosis. Proteome-wide Elucidation of the Molecular Mechanism Defining the Adverse Effect of Selective Esterogen Receptor Modulators. L. Xie and P.E. Bourne 2007 PLoS Comp. Biol., Submitted. Repurposing safe pharmaceuticals to treat multi-drug and extensively drug resistant tuberculosis using an in silico cross-gene-family approach. S. Kinnings, L. Xie and P.E. Bourne 2007 JACS, Submitted. • Ruben Valas • Carnegie Mellon, BS Computer Science 2005 • Bioinformatics Graduate Program • 3rd year PhD student • Rethinking proteasome evolution: • Two novel bacterial proteasomes Feb 1, 2008 Professional Development Series Rethinking proteasome evolution: Two novel bacterial proteasomes. R. Valas and P.E. Bourne 2007 J. Mol. Evol., Submitted.