Download
1 / 43
520 likes | 1.15k Views
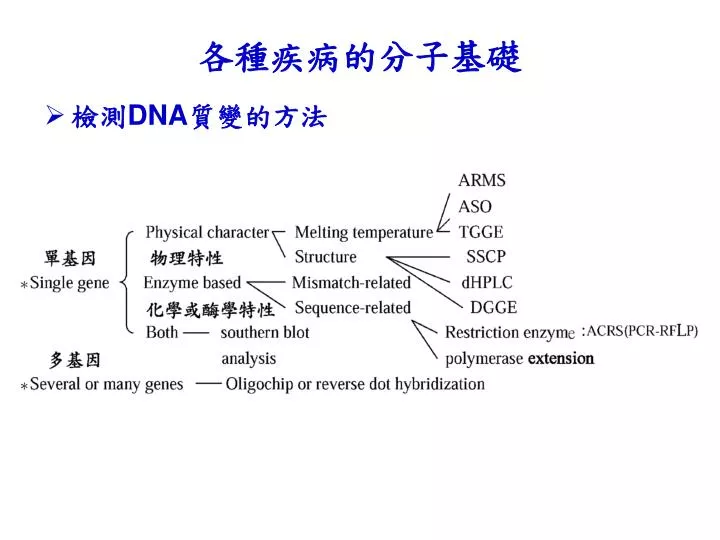
各種疾病的分子基礎. 檢測 DNA 質變的方法. 單基因突變的檢測方法 物理特性 DGGE 變性梯度明膠電泳( denaturing gradient gel electrophoresis PCR 產物跑電泳的分析方法 瓊脂凝膠電 泳:可知大略大小、但長度差異小無法分別 聚丙烯醯胺凝膠電泳:差一兩個鹼基可分開 電泳的速度與片段大小有關,跟 DNA 的組成及序列無關,不能分辨鹼基之間是否有突變 . DNA 雙鏈上發生部分的解鏈,會使電泳速度大大減慢 DNA 被變性的難易程度與 DNA 的長度及組成排列有關 片段長及 CG-rich 需較高的溫度

E N D
各種疾病的分子基礎 • 檢測DNA質變的方法
單基因突變的檢測方法 • 物理特性 • DGGE變性梯度明膠電泳(denaturing gradient gel electrophoresis • PCR產物跑電泳的分析方法 • 瓊脂凝膠電泳:可知大略大小、但長度差異小無法分別 • 聚丙烯醯胺凝膠電泳:差一兩個鹼基可分開 • 電泳的速度與片段大小有關,跟DNA 的組成及序列無關,不能分辨鹼基之間是否有突變
DNA雙鏈上發生部分的解鏈,會使電泳速度大大減慢DNA雙鏈上發生部分的解鏈,會使電泳速度大大減慢 • DNA被變性的難易程度與DNA的長度及組成排列有關 • 片段長及CG-rich需較高的溫度 • DGGE的基本原理 • 雙鏈的DNA部分解鏈成單鏈時,在凝膠電泳速度會大大減慢 • 有鹼基發生突變時,正常與突變跑出來的速度就會不一 樣 • DNA解鏈的溫度還與凝膠中尿素或甲醯胺的濃度有關,濃度會改變電泳速度 • DNA雙鏈的解鏈常是一個小區段接著一個小區段發生,這種小區段稱為變 性區 • 變性區有微小的改變,會改變電泳的速度,藉由電泳的不同將正常與突變檢測出來
變性梯度即尿素或甲醯胺的濃度,從陰極向陽極逐漸增強變性梯度即尿素或甲醯胺的濃度,從陰極向陽極逐漸增強 • 解鏈強度較低的DNA片段先達到其變性梯度而解鏈,電泳速度減慢 • 解鏈強度高的DNA片段保持原來的速度繼續前進,直到達到特定變性梯度區才減慢速度 • 結果將長度相同而序列有差別的DNA片段分離 • 使用溴化乙錠或銀染就 可以觀測區帶的變化 • 設計PCR引子時,擴增片段只含一個變性梯度區 • 引子的5‘端增加20~40 bp左右的CG,即CG clamp,可讓DNA區充分解鏈 • 有時單個鹼基的替換無法改變解離強度 • 將PCR後的兩個檢體產物混合,加熱變性,然後緩慢降溫使形成雜合雙鏈,雜合雙鏈在突變部位形成錯配 • 雜合雙鏈的變性溫度與純合雙鏈有明顯的差別,雜合雙鏈能顯示出突變的存在
一般DGGE使用polyacrylamide gel(含尿素甲醯胺的濃度梯度 • 也可改成溫度梯度,稱為TGGE • 固定變性凝膠電泳CDGE (constant denaturant gel electrophoresis) • DGGE的缺點 • 引子設計複雜 • 必須只有單一的melting domain • 較難控制電泳條件
PCR為基礎的相關技術 • ARMS (Amplified refractory mutation system) • 設計一對Primer可以特異地跟正常或突變的對偶基因結合 • Allele-Specific PCR可用來檢測單鹼基的突變或微小缺失突變 • 缺點:突變與正常的基因型要分開來做, 不能同時在一管中進行 • 1993 Rust et al.建立MS-PCR (Mutagenically separated-PCR) • 在正常(或突變)的引子5'端多加了20 bp • 以Primer長度不同或不同螢光染料來區分 • 靠近3‘端多加一、二個mismatch的鹼基,以增加反應的特異性 • 多個鹼基的改變會使引子與正常或突變序列結合的穩定性不同,這樣就增加引子的特異性。
ACRS (Amplified Created restriction site) • 偵測鹼基變異沒有產生限制酶切位時 • 設計一個與正常有一、三個鹼基差異的mismatch primer 此primer設計在變異的鹼基附近 • PCR後,其產物在primer上的mismatch鹼基及鄰近序列合併變異的鹼基即可產生限制酶的切位 • 設計一個mismatch的primer來產生二種不一樣的PCR 產物,一種產物會被特定的限制酶切,另一種不會被切 • Mismatch base的互補關係較不穩定,設計不佳沒有產物,若溫度調降雜bands多,特異性不足 • 應用ACRS時, Primer的設計就必須特別注意base互補的穩定性。要愈穩定愈好
多重PCR (multiplex PCR) • 當DNA有大段缺損時,利用位於缺損部位的特殊標記,設計引子放大這些標記 • 設計時將每個標記的PCR產物設計成大小不同,且可在同一種PCR狀態進行作用 • 由結果產物的大小變化,即可了解缺損的部分及範圍 • 可用於偵測多個點突變 • 設計多組 Primer ,做PCR反應,將所有可能突變的位點或基因多個缺失熱點區,都擴增分析 • 注意各primer pairs之間應具有相同或相近的擴增條件,相互間不產生非特異性擴增或primer-dimer的情形
各個產物片段長度要適當,必須做 一個正常的內對照組( positive control)以防止擴增的假陰性反應
SSCP單鏈構形多形性single strand conformation polymorphism • 單鏈的DNA在溶液一定的條件下會形成鏈內的局部雙鏈,進而形成特定的立體結構 • 等長的DNA片段,如果鹼基組成及序列的不同就會形成不 一樣的構形 • 特殊的凝膠中電泳,立體構形會影響電泳速度 • 利用已知樣本跑出來的條帶圖形與未知樣本比較,就可以找出待測樣本是否存在突變 • 條帶圖形看起來不太一樣,可將不一樣的樣本做定序,看是否有新的突變位點 • 影響SSCP的因素很多:DNA序列組成與排列,電泳的溫度及凝膠的組成,所分析DNA片段的長度(最好在l50~300 bp左右)
SSCP的結果判定是與正常樣品來比較 • 含未變性徹底的雙鏈、及正鏈和負鏈 • 有時在某些條件下會出現額外的band,可能是某條鏈在這種條件下會形成兩種構形 • 變性不徹底的雙鏈,有同源雙鏈外,還有更多異源雙鏈
SSCP會隨著分析片段長度增加而降低檢出率 • 最好的解析長度是250 bp左右 • 1992年Sarkar等利用Sanger定序法原理,建立雙去氧指紋法( dideoxyl fingerprinting, ddF) • ddF是將雙去氧終止法與SSCP結合的分析技術, • 以一側引子,對PCR產物進行雙去氧末端終止定序反應,以ddCTP加入反應系中, 就會產生一系列長短不一的單鏈DNA,然後再進行SSCP比較分析 • 將 SSCP加上螢光及毛細管電泳的技術就可以提高ddF的靈敏度
異源雜合雙鏈分析Heteroduplex Analysis,HA • HA 在非變性膠電泳上,雜合的雙鏈與純合雙鏈的電泳速度不同,雜合速度會減慢 • 正常基因與突變基因的PCR產物,可以加熱後緩慢冷卻就會形成異源的雜合雙鏈
個體是突變的純合子,則可以將其PCR產物與正常個體的PCR產物 以1:1混合 • 因鹼基錯配,其分子的構形與同源雙鏈DNA不同,可區分開 • HA可分析大於1 kb的片段,但是片段愈大檢測的效率愈差 • 使用溫和變性凝膠電泳,電泳的條件與SSCP類似,這種方法稱為comformation-sensitive gel electrophoresis (CSGE) • 分析200 ~ 800 bp長度DNA片段的多形性
Carbodiimide修飾Carbodiimide Modification • 異源雜合的雙鏈,在C和T的錯配鹼基中,使用Carbodiimide來修飾錯配的地方 • 這種雜合修飾過的錯配雙鏈,在電泳中會改變電泳的速度(被修飾的DNA會跑得比較慢) • 藉由電泳 速度的不同來檢測突變的情形 • 或使用primer做延伸反應,當到了被修飾的mismatch鹼基時就會停止引子的延伸,藉由引子延伸反應的中止而判斷突變的情形
DHPLC變性高效液相層析Denaturing high performance liquid chromatography • 1995年史丹佛大學的Peter Oefner和Umderhill改進 HPLC分析DNA片段的技術 • 分析150 ~1000 bp長度多形性 • HPLC原理:使用高壓幫浦連續按一定流量將 溶劑打入層析性中,再將定量的樣品注入管柱頂端,接著連續洗脫管柱,樣品中各成分,依在管柱中留置的時間( retention time) 不同逐漸分離 • DHPLC也稱為TMHA (temperature-modulated heteroduplex analysis) • 由控制管柱的溫度,選擇一個適當溫度,在這溫度下,雜合的雙鏈在錯配鹼基中會發生部份解鏈,而有部份解鏈的DNA在管柱中結合能力會下降,所以比純合雙鏈更早被洗下。
如果溫度過低沒有發生部份變性的情況,DNA與層析管柱結合的能力取決於DNA的長度如果溫度過低沒有發生部份變性的情況,DNA與層析管柱結合的能力取決於DNA的長度 • 片段越大在管柱中留置的時間就愈長 • 雜合雙鏈產物的產生方法 • 將PCR 產物野生型和突變型以約1 : 1混合 • 95°C加熱5 分, 慢慢降低溫度 • 每一個cycle降1.5°C,共45回 • 會形成雜合雙鏈。然後將8 ml樣本注入管柱 • PCR產物分離條件 • DNA片段的長度及鹼基的組成 • 錯配的雜合子在管柱上的時間會明顯下降 • 先找正常型的DNA片段的實驗條件(管柱的溫度,及緩衝液濃度梯度),得知野生型標準圖譜 • 待測的樣品所跑出來的圖譜與正常的比較 • 建立各種突變的標準圖譜,可藉此快速自動化檢測疾病突變
跑出來的peak有問題,也可以進行直接定序分析跑出來的peak有問題,也可以進行直接定序分析 • DHPLC有許多優點:自動化操作,快速和靈敏度高,應用於單基因突變的篩檢可以在短時間內分析大量的樣品
化學特性 • 錯配鹼基化學切斷法Chemical cleavage of mismatch, CCM • 將篩檢的PCR產物混合,形成異源雜合雙鏈 • 錯配的雜合DNA上的C和T可使用羥胺hydroxylamine及四氧化鋨osmium tetroxide修飾 • 用peperidine切斷DNA的磷酸雙酯鍵 • 切下修飾過的錯配鹼基,依電泳片段的長度大小判斷突變的情形 • 用於1 kb或更長的DNA突變分析上,有極 高的檢出率 • 缺點:須放射性標記及使用有毒的化學物質
錯配鹼基的酶學切斷法Enzyme cleavage of mismatch • T4內切酶VII,會切除錯配的雜合雙鏈 • 單一鹼基的錯配及形成小圈環 ( small loop )都會被切斷 • 可以使用於超過1 kb長度,但有時會有非特異性切 斷產物,使得結果很難分析 • 植物的內切酶CELI效果較好, 較少非特異性的背景產生 • 連接酶鏈鎖反應ligase chain reaction, LCR • 1991 Barany設計二個相鄰的特異性寡核苷酸,只有待測的序列與二個寡核苷酸互補 • 以連接酶將二個相鄰的寡核苷酸連接起來,而有產物產生 • 新的產物又可以在下回合加溫降溫反應中當成新的 模板,經過30回合後,產物跑電泳就可以分辨突變的有無
RFLP (restriction fragment length polymophisms)或PCR-RFLP • 人類非編碼區大約2000~4000 bp中會有一個變異點,編碼區大約1000 bp左右會有一個變異點 • 這些變異是單鹼基(核苷酸)多形性(SNP)的基礎 • 有時突變點會破壞或產生一個限制酶辨識位,這種變化可被DNA blotting或PCR-RFLP檢測出來 • SNP (Single nucleotide polymorphism) • 基因組中特定位點的單一鹼基多形性,非編碼區比編碼區豐富 • 人類基因組中超過三百萬,可以做為基因組的標記 • 這個多形 性的Allele是正常的,或者在某種情況下有潛在致病傾向 • 建構全基因組SNP的晶片,排除個體、種族和環境非疾病因素的影響,使用晶片來尋找致病基因
CFLP (cleavage fragment length polymorphism ) • 單股的DNA在一定條件下會形成鏈內的二級結構,相同長度而序列有變異所形成的二級結構會不相同 • 藉由structure-specific的內切酶 cleavase I來切,就會在正常與變異中形成不同的條帶圖形( band pattern) • Muts • 由E. coli所分泌, Muts蛋白可以跟錯配的DNA結合 • 與錯配的DNA結合,造成錯配的DNA 電泳速度減慢
自動定序Automated sequencing • 使用 Sanger所發明的雙去氧鏈終止法的原理 • 利用DNA聚合酶,一個引子、四種dNTP、四種ddNTP、單股DNA 模板,類似PCR方法, DNA聚合酶催化5‘→3’的複製 • 因ddNTP沒有3‘-OH,不能與後續加入的 dNTP的磷酸基團,形成磷酸雙脂鍵,所以鏈的延伸就會終止 • 調整每個定序反應中ddNTP與dNTP的比例,任何相對的鹼基都有可能被ddNTP所取代,只要一取代就形成鏈的終止 • 定序方法所得到的是大大小單股DNA • 利用4種螢光分別標記4種不同的ddNTPs • 自動定序儀以雷射偵測器,偵測時間和不同螢光顏色, 以電腦加以判讀
Fluorescence dideoxy Method I 參考資料:http://www.cbs.dtu.dk/staff/dave/roanoke/genetics15.htm
一般大量檢體可以先用PCR-RFLP來做或PCR-SSCP分析,有問題的檢 體再送定序 • 檢體量少或很重要的檢體,可以做完PCR後就直接定序 • 所有篩選突變的方法,最終都必須定序來確定 • 定序頭尾比較不正確,最好3‘端及5’端以另一個引子做定序 • 目前技術大概可一次定1000 bp 左右
Minisequencing • PCR時,是引子依據模板逐一加入互補的dNTP做延伸反應 • 此方法則使用ddNTP。所以若在引子的3‘端加入一個雙去氧鹼基就終止了反應 • 加ddNTP的位置就是要突變所在 • 產物跑自動定序分析 • Minisequencing反應產生一個peak就是homozygote (同型合子,純合 子) , 2個peak就是heterozygote (異型合子,雜合子)
初步篩選突變比較常用的是Sequencing , SSCP及DHPLC • 定序是目前最好的方法 • 趨勢是自動化的過程,包括自動化的資料未來分析解讀及自動 化的樣本操作 • 關鍵技術是Microarray Sequencing • 分子診斷只是將DNA序列上可能的異常或突變與正常的比對而已 • 異常及突變太複雜,目前不可能將所有人類的異常突變都點在一片晶片上
